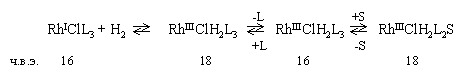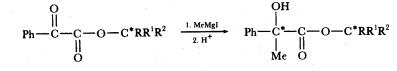Простое гидрирование.
А. Активация водорода путем окислительного присоединения. В 1956 г. две группы исследователей, работающие независимо, сообщили о том, что в органическом растворителе трис(трифенилфосфин)хлорородий(I) RhСl(РРh3)3 является активным катализатором гидрирования алкенов и алкинов при 25 °С и 1 атм газообразного водорода. Вслед за этим было затрачено много усилий для того, чтобы охарактеризовать эту систему. Принятый в настоящее время каталитический цикл показан на рис. 2.1.
Рис. 2.1 Каталитический цикл для гидрирования олефинов в присутствии RhCl3(PPh3). S - растворитель или слабосвязанный третичный фосфин. Внутренний круг представляет собой главный каталитический цикл. Оказалось, что каталитически активные соединения содержат два третичных фосфиновых лиганда, но до сих пор остается открытым вопрос, как такие соединения образуются из трифосфинового комплекса 2.1. Первоначальное предположение о том, что 2.1 быстро диссоциирует в растворе, образуя сольватное соединение 2.3, было до некоторой степени подвергнуто сомнению, когда с использованием измерений молекулярной массы и ЯМР-31Р было показано, что в бензольном растворе в отсутствие кислорода 2.1 практически не диссоциирует (К=3×10-3М). Это полностью соответствует правилу Толмана "16 - 18 электронов" в том отношении, что предполагаемое равновесие подразумевает переход 16-электронного соединения в 14-электронное:
В присутствии подходящего растворителя, способного к координации, например, ЕtOН (при наличии примесей О2), это осложнение может быть преодолено:
В этом смысле примечательно, что скорость гидрирования циклогексена в присутствии катализатора RhСl(РРh3)3 в смеси бензола с этанолом (3:1) примерно в два раза выше скорости реакции в чистом бензоле. Кроме того, на практике трудно полностью исключить кислород. Окислительное присоединение водорода к сольватированному соединению 2.2 приводит к образованию дигидридного комплекса родия(III) 2.3. Данные ЯМР 1Н и 31Р показали, что два гидридных лиганда находятся в цис-положении между собой и в цис-положении к третичным фосфиновым лигандам. Альтернативный путь к сольватированным дигидросоединениям идет через последовательность
Эта последовательность изображена в верхней половине внешнего каталитического цикла на рис. 2.1. Однако несомненно, что формально октаэдрические цис-дигидридные соединения родия(III) активно включаются в каталитический цикл. Гидридные лиганды обладают высоким транс-эффектом; таким образом, транс-лиганд как в 2.3, так и в 2.4 лабилизирован и легко замещается олефиновой группой субстрата, образуя 2.5, в котором все реагенты, необходимые для завершения цикла, собираются вместе в "активированной" форме. Последует ли реакция по внешнему или внутреннему правому полукругу (см. рис. 2.1), будет до некоторой степени зависеть от природы каталитического предшественника, т.е. от комплекса родия, введенного в раствор. Важное преимущество системы на основе RhСl(РРh3)3 состоит в том, что в мягких условиях, обычно при комнатной температуре, в бензольном или бензольно-этанольном растворе при атмосферном давлении водорода эта система не катализирует восстановление других функциональных групп, находящихся в олефиновом субстрате. Такие группы, как кето-, циано-, нитроарильная, карбоксильная и другие обычно встречающиеся органические функциональные группы, не затрагиваются при гидрировании каталитической системой RhСl(РРh3)3/Н2. Этот факт вместе с отмеченными ранее стерическими ограничениями делает RhСl(РРh3)3 исключительно селективным катализатором, имеющим большие возможности в синтезе сложных органических молекул. Тем не менее, прежде чем перейти к более подробному обсуждению этих возможностей, мы должны рассмотреть второй тип процесса активации водорода, а именно гомолитическое присоединение. Б. Активация водорода путем гомолитического присоединения. Впервые об этом типе процесса активации на примере пентацианида кобальта сообщил Игути в 1942 г. Водный или водно-спиртовой (МеОН или EtOH) раствор цианида кобальта при обычных условиях легко поглощает водород, образуя соединения, способные катализировать гидрирование активированных алкенов типа С=С-А По отношению к органическим субстратам эта система высокоселективна в том смысле, что совершенно не реагирует с простыми моноолефинами. Так, сопряженные диены могут быть селективно восстановлены до моноенов, например бутадиен до бутена. Основным соединением, отвечающим за катализ, является анион пентацианогидрида кобальта, который образуется путем гомолитического расщепления водорода:
Использование правила "16 - 18 электронов" в том виде, как оно применялось для диамагнитных комплексов, для этой частной системы ограничено, поскольку СоII(СN)53- - является d7, 17-электронным парамагнитным ионом. Образование гидридокомплекса 2.11 из цианида кобальта и водорода изучалось целым рядом исследовательских групп, и было предложено два механизма, включающих димерные соединения. Однако в большинстве работ отдается предпочтение простой межмолекулярной реакции типа (2.1). Поскольку координированный Н условно принимается за Н-, степень окисления кобальта в 2.11 составляет три. Однако на основании полярографических исследований было предположено, что более правильно полагать, что кобальт в 2.11 присутствует в степени окисления два, а водород присутствует как стабилизированный атом, Это означает, что 2.11 следует представлять как [СоII(СN)5(CН)]3-. Именно это представление соответствовало бы описанию такого процесса активации, как "гомолитическое" присоединение. Но, как будет ясно при рассмотрении каталитического цикла, более важно то, что это представление хорошо согласуется с природой реакций, протекающих с участием НСо(СN)53-. Для того чтобы в дальнейшем избежать путаницы с ранее предложенным формальным определением гомолитического присоединения, мы в последующем обсуждении воздержимся от написания степени окисления кобальта. Каталитический цикл для гидрирования активированных олефинов, катализируемых НСо(СN)53- показан на рис. 2.2. Можно представить, что начальное взаимодействие 2.11 с активированным олефином происходит через четырехцентровое переходное состояние 2.12, в котором атом водорода перемещается к активированному группой А b-атому углерода. Каталитический цикл, которому теперь будет следовать система, зависит от природы активированного олефина. Сопряженные диены и полиены следуют циклу (I), тогда как большинство других активированных субстратов гидрируется по циклу (II). Ниже мы рассматриваем гидрирование двух характерных субстратов - бутадиена и коричной кислоты.
Гидрирование по циклу (1) на примере бутадиена. При взаимодействии с сопряженными диенами, такими как бутадиен-1,3, алкилкобальтовое соединение типа 2.13 формально образуется путем цис-присоединения гидрида кобальта к активированной двойной связи. Природа моноолефинового продукта, образующегося по циклу (1), зависит от концентрации свободного цианида, присутствующего в реакционной среде. В случае бутадиена, когда гидрирование проводится в присутствии избытка цианида, т.е. когда отношение цианид/кобальт выше чем 5:1, основным продуктом (~90%) является бутен-1. В присутствии небольшого количества свободного цианида (<5) основным продуктом (~80%) является транс-бутен-2. В обоих случаях образуется лишь небольшое количество (~5%) цис-бутена-2. Эту зависимость продуктов от концентрации цианида можно объяснить, использую последовательность, показанную на рис.2.3.
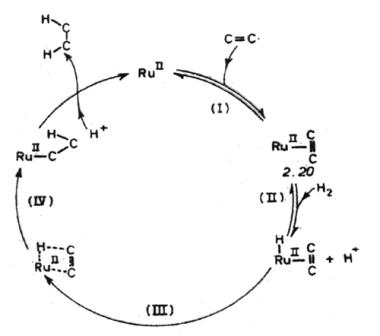
Присоединение CoH(CN)53- к бутадиену через 2.12 (А= -СН=СН2) приводит к образованию s-комплекса 2.14. Потеря цианида комплексом 2.14 создает вакантное координационное место в октаэдрическом комплексе, координированная двойная связь начинает взаимодействовать с этим вакантным координационным местом, и фрагмент С4, занимающий одно координационное место, превращается из s-лиганда в p-связывающий лиганд, занимающий два координационных места в комплексе 2.15. Метильная группа в этом комплексе направлена в противоположную сторону от кобальта, т.е. находится в син-положении. Этот тип s-p-превращений, широко распространенный среди комплексов металлов VIII группы, содержащих аллильную группу, широко будет рассмотрен при рассмотрении изомеризации. Присоединение CN- к 2.15 может привести к образованию либо снова 2.14, либо 2.15, в котором кобальт теперь s-связан с С=4. Хотя детальный механизм, по которому CoH(CN)53- взаимодействует с s- и p-соединениями 2.14 и 2.16, неизвестен, подробные спектроскопические и кинетические исследования показали, что транс-бутен-2 предпочтительно образуется из p-аллильного комплекса 2.15, а бутен-1 - из s-комплекса. В реакционных условиях в обоих случаях присутствует лишь небольшое количество 2.14, но именно этот комплекс, вероятно, и приводит к образованию бутена-1. Очевидно, что низкая концентрация цианида будет благоприятствовать образованию и, что очень важно для распределения продуктов, присутствию заметных количеств 2.15. Это согласуется с тем, что при отношении CN:Co менее 5:1 в основном образуется транс-бутен-2. Эта система высокоселективна в восстановлении бутадиена, поскольку дальнейшего гидрирования или изомеризации получающихся бутенов не происходит. В рассмотренном выше случае все предполагаемые интермедиаты - кобальторганические соединения, в которых субстрат ковалентно связан с кобальтом. Для кобальтцианидного катализатора гидрирования это справедливо только в том случае, когда субстратом является сопряженный диен или полиен. Как отмечалось ранее, в случае любых других активированных субстратов интермедиатами являются свободные радикалы, как это показано в цикле II (рис.2.2). В. Активация водорода путем гетеролитического присоединения. Водные солянокислые растворы хлорида рутения(II) катализируют гидрирование a-ненасыщенных карбоновых кислот или амидов, например малеиновой, фумаровой, акриловой кислот или акриламида, однако они неактивны в восстановлении простых олефинов и большинства других органических субстратов. Несмотря на то что еще не ясна природа хлоридного комплекса рутения(II), активного в каталитическом цикле, вполне разумно предположить, что активация водорода происходит путем гетеролитического присоединения к олефиновому комплексу рутения(II)., Каталитический цикл, согласующийся с известными в настоящее время экспериментальными данными, показан на рис. 2.4.
Рис. 2.4 Каталитический цикл гидрирования активированных олефинов в водных растворах соляной кислоты, катализируемого комплексами рутения (II). На этом рисунке не делается никаких попыток точно установить природу соединений рутения(II). Присутствие заместителя, сильно оттягивающего электронную плотность, например - СООН или - СОNН2 необходимо для активации алкена. Незамещенные олефины, такие, как этилен или пропилен, в водной соляной кислоте легко образуют комплексы 2.20 с рутением(II). Однако эти комплексы относительно стабильны и не гидрируются. Предполагается, что стадия II в каталитическом цикле обратима и что миграция лиганда (Н к олефину) в стадиях III и IV может происходить только тогда, когда олефин сильно активирован для нуклеофильной атаки. В этой системе, в противоположность рассмотренной ранее кобальтовой системе, очень вероятно, что лиганд Н реагирует как Н-. Изучение дейтерирования показало, что присоединение Н2 к олефину происходит стереоспецифически в цис-положение. Это указывает на то, что электрофильная атака Н+ рутенийалкильного соединения происходит с сохранением конфигурации углеродного атома, связанного с атомом металла. К сожалению, из-за экспериментальных сложностей, возникающих при работе с этой специфической системой, оказалось трудно охарактеризовать большинство индивидуальных промежуточных соединений, и поэтому подробности каталитического цикла остаются в значительной мере невыясненными. Другой системой на основе рутения(II), в которой существенную роль играет гетеролитическое присоединение водорода, является система, образующаяся при реакции комплекса рутения(II) типа RuСl2(РРh3)n (n = 3 или 4) с водородом при комнатной температуре в бензольно-этанольном растворе. Образующийся гидридокомплекс RuСl(H)(РРh3)3 является одним из наиболее активных из до сих пор найденных гомогенных катализаторов гидрирования алкенов-1. Он также катализирует восстановление нитросоединений в амины и альдегидов в спирты. Несмотря на то что эта система по сравнению с системой RuСl2/НСl/Н2О намного легче поддается изучению, например, методом ЯМР - 1Н и 31Р, детали каталитического цикла участием RuСl(Н)(РРh3)3 остаются до некоторой степени еще невыясненными. Цикл, представленный на рис.2.5, служит просто для объяснения известных в настоящее время данных.
Рис. 2.5 Каталитический цикл для гидрирования олефинов в присутствии RuIICl(H)PPh3)3. Селективное гидрирование. В настоящее время известны гомогенные системы, способные при относительно мягких условиях катализировать восстановление большинства обычно встречающихся ненасыщенных органических групп. Однако именно селективность таких систем, т.е. их способность восстанавливать только один определенный вид ненасыщенности в присутствии других чувствительных или потенциально восстанавливаемых видов ненасыщенности, а не их универсальность делает гомогенные каталитические системы исключительно полезными в синтетической органической химии. Мы уже кратко затрагивали этот аспект при обсуждении системы RhСl(РРh3)3. Далее мы несколько более подробно рассмотрим некоторые типы возможных селективных превращений. А. Гидрирование простых алкенов и алкинов. Гомогенными катализаторами, способными селективно восстанавливать алкены и алкины в присутствии других функциональных групп, являются RhСl(РРh3)3, RuСl2(РРh3)3 и RhН(СО)(РРh3)3. В мягких условиях - при 25 °С и давлении водорода 1 атм - все три катализатора являются очень высокоселективными по отношению к алкеновой или алкиновой функциональным группам, причем другие ненасыщенные группы, такие, как -СНО, -С-СО2Н, -СN или -NО2, не затрагиваются. Путем тщательного выбора условий реакции можно селективно восстановить алкин в соответствующий алкен в присутствии другого алкина и алкена. Так, в фенолбензольном (1:1) растворителе RhСl(РРh3)3 катализирует восстановление смеси 1:1 гексина-1 и октена-1 таким образом, что только после полного восстановления гексина-1 начинает восстанавливаться либо октен-1, либо продукт гексен-1. Хотя абсолютные скорости гидрирования алкина обычно ниже скоростей гидрирования соответствующих алкенов, в смеси алкен-алкин обычно более предпочтительно восстанавливается алкиновая функциональная группа благодаря ее более высокой способности образовывать связь с металлическим центром. Все три каталитические системы исключительно чувствительны к стерическим изменениям в субстрате и отдают определенное предпочтение терминальным алкенам или алкинам. Это особенно заметно для RuСl2(РРh3)3 - самой активной из трех указанных систем, в присутствии которой олефины с концевой двойной связью гидрируются примерно в 10 000 раз быстрее, чем соответствующие внутренние олефины. RhСl2(РРh3)3 слегка менее селективен, чем RuС1(РРh3)3, хотя он еще предпочтительнее гидрирует терминальные алкены: скорость гидрирования октена-2 в четыре раза ниже скорости гидрирования октена-1. Скорость восстановления имеет следующую тенденцию: алкен-1 > цис-алкен-2 >> транс-алкен2 > транс-алкен-3. Б. Гидрирование сопряженных диенов, полимеров и активированных полимеров. Каталитическая система на основе раствора цианида кобальта в водной соляной кислоте является высокоспецифичной по отношению к гидрированию сопряженных диенов, полиенов и отдельных активированных мономеров. Однако в действительности эта система имеет один недостаток, связанный с тем, что ее обычно приготавливают в водном растворе, в котором большинство органических субстратов слабо растворимо. Другие гомогенные катализаторы, селективно восстанавливающие диены в моноены, обычно менее активны, однако три каталитические системы - на основе Fe(CO)5, циклопентадиениловые комплексы хрома, молибдена или вольфрама, и хлористооловянно-фосфиновые комплексы платины могут успешно работать в органических растворителях. Обычно реакцию проводят при температуре выше 100 °С и давлении водорода порядка 30 атм. В. Гидрирование ароматических и гетероциклических соединений. Большинство гомогенных каталитических систем неэффективно в восстановлении ароматических углеводородов. Промышленное применение имеют некоторые цигглеровские системы, т.е. комбинации солей переходных металлов, обычно галогенидов, с алкилалюминием или с алкилалюминийхлоридом. Асимметрическое гидрирование. Асимметрическим синтезом является процесс, в котором ахиральное соединение или совокупность таких соединений превращается в хиральное таким образом, что образующиеся стереоизомерные продукты или энантиомеры получаются и неодинаковых количествах. Эффективность асимметрического синтеза измеряется величиной преобладания (избытка) одного энантиомера над другим, выраженной в процентах, т.е. процентом энантиомерного избытка (%э.и. =%R-%S). Процент энантиомерного избытка - это синоним обычно более часто используемого названия "оптическая чистота". Асимметрический синтез является исключительно важным в фармацевтической и агрохимической промышленности, поскольку зачастую два оптических изомера одной и той же молекулы имеют заметную разницу в фармакологической и биологической активности. Имеются два способа осуществления асимметрического синтеза. Первый из них - создание второго хирального центра под влиянием присутствующего в молекуле хирального центра, например:
Другой путь - создание нового хирального центра в прохиральном субстрате под действием хирального реагента или катализатора. Асимметрическое гидрирование является наиболее широко распространенным примером второго способа проведения асимметрического синтеза с участием катализатора. Если хиральным катализатором является комплекс переходного металла, то хиральность может быть введена в основном двумя способами: либо сам металл, если он окружен различными лигандами, может быть центром хиральности, либо хиральными могут быть один или несколько лигандов, связанных с металлом. В случае асимметрического гидрирования более важным является именно второй способ. Почти все известные в настоящее время гомогенные катализаторы асимметрического гидрирования основаны на комплексах родия с третичными фосфиновыми лигандами. В настоящее время приняты два подхода к введению оптической активности в такие системы, а именно использование третичных фосфиновых лигандов с асимметрическим фосфором типа PRR1R2 или использование лигандов, содержащих асимметричный атом в одном из заместителей при атоме фосфора, например Р*R*R1R2, где P* = Рh или R*- ментил или неоментил. Гидрирование путем переноса водорода. Все реакции гидрирования, которые мы обсуждали до настоящего времени, были реакциями типа Поскольку имеется много общего между гидрированием путем переноса водорода и гидрирование с участием молекулярного водорода, то большинство из обсуждавшихся ранее каталитических систем активны и в гидрировании путем переноса водорода. Основным отличием между гидрированием собственно водородом и путем переноса водорода является то, что в случае гидрирования путем переноса водорода не происходит изменения общей ненасыщенности в системе; в простейшей форме это выглядит следующим образом: "алкан (1)" + алкен (1) ® алкен (2) + "алкан (2)" Это накладывает определенные ограничения на реакции гидрирования путем переноса водорода. Донор водорода "алкан (1)" должен не только обладать способностью взаимодействовать с металлическим центром с образованием по крайней мере одного атома водорода, но также в разумных пределах конкурировать с алкеном (1) за вакантное координационное место катализатора. Практически это означает, что молекула донора должна содержать функциональную группу (напр. донорный кислород или азот), способный координироваться с металлическим центром. Кроме того, образовавшийся продукт дегидрирования донора (алкен (2)) должен покидать координационную сферу, освобождая место для акцептора водорода (алкена (1)). Эти ограничения проявляются в сложной кинетике таких систем обычно происходит ингибирование реакций продуктом (алкеном (2)). Гидрирование путем переноса водорода является, по существу, реакцией образования двух продуктов, и поэтому оно менее привлекательно с практической и коммерческой точки зрения. Тем не менее оно может быть полезным, если, например учесть большую безопасность по сравнению с процессом в присутствии газообразного водорода или если в используемой реакционной среде уже присутствуют подходящие доноры и акцепторы, например в нефтяной фракции, получаемой в процессах нефтепереработки и риформинга. Однако в последнем случае гетерогенная каталитическая система более предпочтительна, чем гомогенная.
|