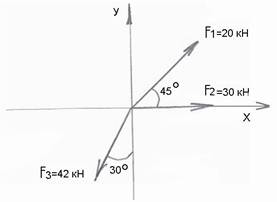
Минимальная мозговая дисфункция (ММД, синдром гиперактивности, гипермоторного ребенка)
Развитие данного синдрома связано с незрелостью и снижением активности тормозных механизмов головного мозга. Поэтому, в некоторых зарубежных странах, для лечения этого синдрома используют амфетамины, запрещенные к применению в России (препараты попадают в категорию наркотических веществ, вызывающих быстрое привыкание). Так же используются различные элементы педагогической коррекции, занятия с психологом и логопедом, упражнения на концентрацию внимания. 8.Наследственные заболевания с преимущественным поражением пирамидной и мозжечковой систем (системная спастическая параплегия Штрюмпеля, семейная атаксия Фридрейха, наследственная мозжечковая атаксия Пьера-Мари). Медленно прогрессирующие заболевания с благоприятным в отношении жизни прогнозом. Трудоспособность больных ограничена, а на более поздних этапах утрачена. 8.1 Болезнь Фридрейха – самая частая форма наследственных атаксий (2-7 на 100 тыс. населения), наследуется по аутосомно-доминантному типу. Ген болезни картирован, что дало возможность проводить раннюю и пренатальную ДНК-диагностику заболевания и определять гетерозиготное носительство мутантного гена. Первые симптомы заболевания проявляются на 1-2-м десятилетии жизни вследствие медленно прогрессирующей демиелинизации боковых и задних столбов спинного мозга с преимущественным поражением мозжечковых и пирамидных путей. Болезнь проявляется расстройствами координации движений, нарушениями глубокой чувствительности, парапарезом. Отмечаются скелетные деформации («стопа Фридрейха», кифоз, кифосколиоз, деформации пальцев); эндокринные расстройства (сахарный диабет, гипогонадизм, инфантилизм, дисфункция яичников); кардиомиопатии; катаракта; снижение интеллекта. Мужчины болеют чаще женщин. Отмечается картина спастической параплегии с заметным преобладанием мышечной гипертонии. В положении лежа спастичность в ногах значительно уменьшается. У больных иногда отмечаются слабо выраженные расстройства чувствительности, атаксия, нистагм, деформация стоп, небольшие дистальные мышечные гипотрофии, императивные позывы к мочеиспусканию. Из наследственных болезней, обусловленных преимущественным поражением экстрапирамидной системы, наибольший интерес представляют хорея Гентингтона, гепатоцеребральная дистрофия (болезнь Вильсона-Коновалова), торсионная дистония и миоклонус-эпилепсия. Хорея Гентингтона – хроническая прогрессирующая атрофия клеток хвостатого ядра и полшарий большого мозга, проявляющаяся распространенным хореическим гиперкинезом, деменцией и неуклонным прогрессирующим течением. Болезнь наследуется по аутосомно-рецессивному типу и возникает у лиц старше 35 лет. Гепатоцеребральная дистрофия наследуется по аутосомно-рецессивному типу и характеризуется прогрессирующим нарастанием сочетанного поражения головного мозга и печени, в основе которого лежит снижение синтеза церулоплазмина и как следствие – нарушение обмена меди. Наиболее типичны две клинические формы заболевания: дрожательно-ригидная и гиперкинетическая. Дрожательно-ригидная форма начинается в возрасте от 15 до 25 лет. Характерно развитие ригидности и тремора, возможно возникновение дизартрии и дисфагии. Тремор усиливается при целенаправленных движениях. Без лечения заболевание прогрессирует в течение 5-6 лет и заканчивается гибелью больного. Гиперкинетическая форма отличается более доброкачественным течением, начинается в возрасте 20-25 лет, а иногда и значительно позже. У больных развиваются гиперкинезы – хореоатетоз, тонические судороги, гемибаллизм. Гепатоцеребральная дистрофия сопровождается изменением психики в виде аффективных расстройств, а на поздних этапах – развитием слабоумия различной степени. Печень поражается до развития неврологической симптоматики. Вначале возникает гепатоз или гепатит, при длительном течении заболевания – цирроз печени, затем портальная гипертензия с повторными кровоизлияниями из варикозно расширенных вен пищевода. Кровоточивость десен, носовые кровотечения могут задолго предшествовать развитию неврологических проявлений. Патология печени достаточно быстро приводит к нарушению метаболизма эстрогенов и других гормонов. Уже на ранних этапах болезни для женщин характерны нарушения кальциевого и фосфорного обмена и как следствие – спонтанные переломы костей, разрушение зубов, деформирующий артроз, возникновение гломерулонефрита, мочекаменной болезни и солевого цистита. Заболевание характеризуется частой аменореей, самопроизвольными абортами. Для лечения больных с гепатоцеребральной дегенерацией повсеместно используют D-пеницилламин – тиоловый препарат, образующий комплексы с тяжелыми металлами. Торсионная дистония чаще всего манифестирует в возрасте до 20 лет, делится на генерализованные и локальные (блефароспазм, тризм, лицевой параспазм, кривошея, «писчий спазм» и др.) формы, кроме того, на ригидные и гиперкинетические формы. При ригидных формах очень хороший эффект оказывает использование низких доз леводопы. Возможно сочетание приема дофасодержащих препаратов с бромокриптином и селегимином. При гиперкинетических формах возможно использование диазепинов (клоназепам). Среди наследственных болезней обмена, протекающих с поражением нервной системы, для неврологии акушерства интерес представляют наследственные заболевания соединительной ткани – болезнь Марфана, синдром Черногубова-Элерса-Данлоса, синдром Ван дер Хаве, или синдром голубых склер. У этих заболеваний немало общих черт – деформация скелета, избыточная мобильность суставов с привычными вывихами, гиперрастяжимость связок и кожи, повышенная ломкость костей и сосудов, пороки развития сосудов (стеноз легочной артерии, коарктация аорты, аневризмы), пролапс митрального клапана, мышечные дистрофии, имитирующие миопатический синдром, патология зрительного аппарата, врожденные пупочные, паховые грыжи, выпадения прямой кишки, матки, дивертикулы желудочно-кишечного тракта, мочевого пузыря и др. Профилактика наследственных заболеваний осуществляется путем проспективного (до планирования деторождения) и ретроспективного (после рождения больного ребенка) медико-генетического консультирования. Безусловными показаниями для направления на консультацию к врачу-генетику можно считать повторные выкидыши, мертворождения, наследственные болезни в семье, браки между близкими родственниками, появление в семье ребенка с врожденными пороками, задержкой физического и умственного развития при вероятном тератогенном воздействии в первом триместре беременности. При высоком риске рождения больного ребенка после тщательной пренатальной диагностики рекомендуется прерывание беременности. Концентрация АФП понижена в крови женщин, вынашивающих плод с болезнью Дауна, повышена при врожденных дефектах нервной трубки. Повышение содержания белка в крови требует его исследования в амниотической жидкости и тщательного ультразвукового исследования, понижение концентрации АФП предполагает цитогенетическое исследование клеток (амниоцитов или лимфоцитов плода). Исследование уровня хорионического гонадотропина позволяет заподозрить наличие у плода хромосомной аберрации. Вышеописанные мероприятия относят к первичной профилактике наследственной патологии. К вторичной профилактике можно отнести использование специальных диет при фенилпировиноградной олигофрении, галактоземии, прием фолиевой кислоты до зачатия и в течение первых 3 месяцев беременности – при повышенном риске развития дефектов нервной трубки плода (сокращает этот риск на 65-70%).
|