Влияние опухоли на организм
Влияние опухоли на организм 6. Механизмы антибластомной резистентности 7. Иммунотерапия и генная терапия опухолей.
Общая характеристика опухолевого роста, определение. С1Опухолевый процесс – динамично развивающийся типовой патологический процесс, который возникает под действием канцерогенных веществ, характеризуется патологическим разрастанием структурных элементов тканей, атипизмом роста, обмена веществ, структуры и функции опухолевых клеток и тканей и приводит к нарушению жизнедеятельности всего организма. С2ОП формируется на основе особой формы нарушения тканевого роста – опухолевого роста. Существуют изменения тканевого роста, такие как гипертрофия, гиперплазия, гипоплазия, которые имеют адаптивный характер, жестко контролируются многообразными регулирующими системами организма и приостанавливаются, когда масса и / или число клеток обеспечивает уровень функции клетки, органа или системы, соответствующий потребностям организма. При повреждении генетического контроля размножения и созревания клеток, их рост приобретает патологический характер – в форме опухоли, которая не только не соответствует потребностям организма, но и приводит к тяжелым нарушениям обмена веществ, структуры и функции других клеток, органов и систем, к изменениям гомеостаза, нарушениям жизнедеятельности всего организма и формированию опухолевой болезни с той или иной локализацией. С3Опухолевый процесс широко распространен среди живых существ, как по вертикали, так и по горизонтали, т.е. обнаруживается у организмов всех уровней эволюции, а среди представителя одного вида (в частности человека) имеется у лиц разного возраста, пола, конституции и в различных органах и тканях. Злокачественные опухоли как причина смерти находятся на втором месте (20% общей смертности) после сердечно-сосудистых заболеваний.
II. Этиология опухолей. Причиной опухолей могут быть разнообразные факторы физической, химической и биологической природы, обладающие мутагенной активностью, т.е. способные вызывать такие изменения генетического аппарата соматической клетки, которые трансформируют фенотип и передаются по наследству от материнской к дочерним клеткам при их размножении. 1. Физические канцерогены: - ионизирующая радиация, -УФ излучение, -термические факторы (длительно действующие), -механические факторы (травмы, ушибы) 2. Химические канцерогены: -экзогенные – органические и неорганические вещества, являющиеся продуктами социальной деятельности человека, в том числе некоторые лекарственные соединения, полициклические ароматические углеводороды (ПАУ), нитрозамины, причем сами по себе потенциальные канцерогенные вещества не вызывают ОР. Поэтому их называют про- или преканцерогенами. В организме они подвергаются физико-химическим превращениям, в результате которых становятся истинными, конечными канцерогенами; -эндогенные – образуются в самом организме: индол, эстрогены, желчные кислоты. 3. Канцерогенные биологические факторы: - микотоксины – токсины плесневых грибов, которые при наступлении в организме вызывают опухоли (например, афлотоксин Aspergillus flavus на арахисе) -вирусы (ДНК- и, особенно, РНК-содержащие). К онкогенным ДНК – содержащим вирусам относятся: - а) папова – вирусы, - б) аденовирусы (12,18,31–го типов), - в) герпес-вирусы (вирус Эпштейна – Бар вызывает лимфому Беркита, вирус гепатита B – рак печени, вирус папилломы человека – рак шейки матки). ДНК вирусов непосредственно встраивается в геном клетки-мишени, вызывая ее мутацию и трансформацию в опухолевую клетку. Вместе с тем ДНК- содержащие вирусы чаще вызывают инфекционные процессы, т.к. после воспроизведения вирусных частиц клетки–мишени гибнут. Большее значение в онтогенезе имеют РНК–содержащие вирусы, являющиеся ретровирусами. После инфицирования клетки, с помощью фермента ревертазы (обратная транспритаза) происходит синтез 2-спиральной ДНК на матрице односпиральной РНК. Образуется ДНК- копия вируса, которая затем может встраиваться в геном клетки-хозяина. В зависимости от онкогенности ретровирусы разделяют на две группы: -остротрансформирующие ретровирусы – очень онкогенны, вызывают развитие опухолей после короткого латентного периода. Эти вирусы имеют в своем геноме онкоген (вирусы острых лейкозов у птиц, мышей, саркома Рауса у кур); -медленно трансформирующие вирусы – вызывают развитие опухолей после длительного латентного периода. Они не имеют в своем составе онкогена, поэтому основной механизм их действия – мутационный (вирусы лимфолейкоза). В возникновении опухолевого процесса важное значение имеет реактивность организма, в частности: -видовая (вирус Рауса вызывает саркому у кур, а вирус Эпштейна-Бар – лимфому Беркита у человека). -групповая (наиболее часто опухоли возникают в возрасте старше 50 лет и у мужчин в 1,5-2 раза чаще, чем у женщин) -индивидуальная, которая определяется состоянием нервной системы, обусловливающей реакцию организма на стрессорные факторы, эндокринной и, особенно, иммунной системы. Важное значение в возникновении опухолей имеет наследственная предрасположенность. Некоторые опухоли у детей (ретинобластома, гепатобластома) являются наследственными заболеваниями. Однако чаще наследуется предрасположенность к возникновению опухолей, например при пигментной ксеродерме вероятность возникновения рака кожи возрастает в 1000 раз, при атаксии-телеангиэктазии и анемии Фанкони повышается частота возникновения лейкозов.
III. Патогенез опухолей. 1.Онкогены и протоонкогены – определение, общая характеристика. Нормальные гены, участвующие в переносе митогенного сигнала называются протоонкогенами. Протоонкогены — это собственные гены клеток, которые несут информацию о структуре белков, принимающих участие в регуляции клеточного деления. Протоонкогены есть во всех клетках. Однако в одних клетках они всю жизнь репрессированы, т.е. "молчат", в других они "работают" только в период эмбриогенеза, а в остальных — функционируют в соответствии с поступающими регуляторными сигналами. Протоонкогены являются клеточными аналогами вирусных онкогенов. Считают, что вирусные онкогены представляют собой протоонкогены, попавшие в геном вирусов в результате длительной эволюции последних. Это объясняется тем, что вирусы, проходя через клетки, могут "воровать", т.е. прихватывать с собой клеточные гены.
Исходным положением в современной концепции онкогенеза является то, что различные канцерогенные воздействия при определенных условиях прямо или опосредованно вызывают мутацию соматических клеток, т.е. изменяют свойства определенных структур ДНК, контролирующих деление и созревание клеток (см. рисунок механизмы канцерогенеза). Так, при действии остро трансформирующих вирусов, сам вирусный онкоген вызывает опухолевую трансформацию клетки - т.н. эпигеномный механизм опухолевой трансформации. С другой стороны под действием физических, химических факторов и вирусов, не содержащих онкогены возможна активация собственных протоонкогенов, они превращаются в онкогены и вызывают опухолевую трансформацию. Т.о. онкогены не являются специфическими компонентами лишь одних вирусов, а существуют в норме во всех клетках организма в форме неактивных протоонкогенов и при стимуляции, превращаясь в онкогены, усиливают митотическое деление клеток. Протоонкогены обнаружены в составе ДНК клеток практически всех живых существ от одноклеточных до животных и человека. Вирусы являются переносчиками протоонкогенов, которые, находясь в составе вирусов, меняют свои свойства, превращаясь в онкогены. В настоящее время обнаружено большое число онкогенов, (более 20) которые обеспечивают образование различных онкобелков с различными функциями. Эти онкогены обозначают соответственно по нахождению – в вирусе (v-virus) или в клетке (c-cellule), а также по характеру опухоли; например: v-src-вирус саркома Рауса, v-sis – вирус саркомы обезьян (simian – обезьяна), c-myc – клеточный онкоген миелоцитарного лейкоза, c-erb – клеточный онкоген эритробластоза, c - myb – клеточный онкоген миелобластного лейкоза. Онкогены кодируют все белки (онкобелки), ответственные за вовлечение клетки в процесс деления. В зависимости от продуктов, информацию о которых несут онкогены вирусов и протоонкогены клеток, их делят на следующие группы (рис. 56).
1). Онкогены, кодирующие факторы роста или их аналоги. Ростовые факторы (регуляторы пролиферации)- это небольшие белки, которые секретируются одними клетками и действуют паракринным образом на другие. Существует несколько семейств ростовых факторов, члены каждого из которых объединены структурной гомологией и функциональным сходством. Одни из них стимулируют пролиферацию (например, эпидермальный фактор роста -ЭФР и тромбоцитарный фактор роста –ТцФР), а другие (ФНО, интерфероны) — ее подавляют. Примером онкогена, кодирующего факторы роста является sis-онкоген вируса саркомы обезьян и аналогичный протоонкоген клеток, кодирующих структуру тромбоцитарного фактора роста (ТцФР). 2). Онкогены, кодирующие клеточные рецепторы факторов роста. Рецепторы к ростовым факторам расположены на клеточной поверхности. Каждая клетка обладает своим особым репертуаром рецепторов и, соответственно, своим особым набором ответных реакций. Тирозинкиназные рецепторы (ТКР) состоят из нескольких доменов: внеклеточного (взаимодействующего с лигандом), трансмембранного и подмембранного, обладающего тирозинпротеинкиназной активностью (см. рис.56 - 3). При связывании с ростовыми факторами (например, ЭФР) молекулы рецепторов инициируют реакции, вследствие чего возникает трансмембранный перенос сигнала — зарождение той волны «возбуждения», которая распространяется затем в виде каскада реакций фосфорилирования внутрь клетки и благодаря которой митогенный стимул достигает в конце концов генетического аппарата ядра. Примерами онкогенов, кодирующих клеточные рецепторы факторов роста, является онкоген вируса лейкоза кур erb-B, он несет информацию о видоизмененном рецепторе к эпидермальному фактору роста (ЭФР), и scr-онкоген вируса саркомы Рауса и соответствующий протоонкоген клеток, кодирующие тирозинспецифические протеинкиназы. 3). Онкогены, кодирующие структуру белков, переносящих информацию от рецепторов плазматической мембраны клетки к ядру. Одним из наиболее важных является сигнальный путь с участием Ras-белков (это подсемейство т.н. G белков, образующих комплексы с гуаниловыми нуклеотидами; Ras-GTP — активная форма, Ras-GDP — неактивная). Он опосредует разнообразные сигналы, исходящие из внешней среды, и функционирует, по всей вероятности, в каждой клетке организма. Ras-белки играют роль своеобразного турникета, через который должен пройти почти любой из поступающих в клетку сигналов. Критическая роль этого белка в регуляции клеточного деления известна с середины 80-х годов, когда активированная форма соответствующего гена (онкоген ras') была обнаружена во многих опухолях человека. Примерно 30% всех опухолей человека содержат мутантные варианты белков ras. Причем для карцином толстой кишки, поджелудочной железы и щитовидной железы этот показатель еще выше. Ras-опосредованный сигнальный путь контролирует т.н. MAP (mitogen achvated ргоtein)-киназный каскад, т.е. каскад реакций фосфорилирования, возникающий как следствие митогенной активации клетки. Активность ферментов, участвующих в киназных каскадах, уравновешивается активностью противодействующих им и находящихся под столь же строгим контролем фосфатаз. Результатом активации МАР-киназ является индукция ряда факторов транскрипции и, как следствие, стимуляции активности ряда генов. 4). Онкогены, кодирующие структуру ядерных белков-регуляторов. Все системы и пути сигнальной передачи замыкаются в ядре клетки и воздействуют на отвечающие гены, которые организуют продвижение клетки через митотический цикл. Репликация ДНК и деление клетки регулируются семейством генов, продукты которых локализуются в самом ядре, где они контролируют транскрипцию генов, связанных с ростом. Неудивительно, что эти гены ассоциируются со злокачественной трансформацией. Известна внутриядерная локализация множества онкобелков, производных онкогенов myc, myb, jun и fos.
Рис.2. «Рефлекторная дуга» митогенного сигнала. Нарушение регуляции экспрессии гена с-myc встречается при лимфоме Беркита, В-клеточных опухолях, тогда как усиление N-myc и L-myc – в нейробластомах и мелкоклеточном раке лёгкого (Рис.3.). Продукты вирусных онкогенов являются аналогичными продуктам протоонкогенов, т.е. собственным белкам клеток, принимающим участие в регуляции клеточного деления. Однако есть и некоторые отличия, связанные с различиями в структуре вирусных онкогенов и протоонкогенов. Именно эти различия делают продукты вирусных онкогенов собственно онкогенными для клетки. Выделяют несколько механизмов трансформации протоонкогена в онкоген: 1). Активация протоонкогена клетки при включении в ее геном вирусного промотора – инсерционная активация. 2). Амплификация – увеличение числа одинаковых копий онкогена, что приводит к возрастанию синтеза онкобелков. При этом могут появляться добавочные участки хромосом. 3). Транслокация участка хромосом в другое место той же или другой хромосомы, что приводит к активации протоонкогена (Ph-хромосома). 4). Точечная мутация протоонкогена, приводящая к его активации (экспрессии). 5). Гипометилирование – уменьшение метилирования ДНК, 5-метилцитозин заменяется на тимин, что является мутацией и может активировать протоонкоген. Гипометилирование наблюдается при старении организма, сопровождаясь возрастанием частоты возникновения опухоли. К онкогенам относят также гены, ответственные за такие свойства злокачественных клеток как: иммортальность, инвазивный рост, тумурогенность, метастазирование. Эти онкогены дополняют друг друга, т.е. являются комплементарными. Их включение происходит независимо друг от друга и неодномоментно и обусловливает опухолевую прогрессию.
2. Гены – супрессоры (антионкогены) и их роль в делении клетки. Баланс позитивных и негативных факторов — фундаментальное свойство любой сложной регуляторной системы, в том числе и управляющей клеточным делением. Протоонкогены — элементы позитивной регуляции; они являются акселераторами клеточного деления и в случае превращения в онкогены проявляют себя как доминантный признак. Вместе с тем, в давних опытах по образованию гетерокарионов (продуктов слияния клеток в культуре) установлено, что свойство туморогенности (способности образовывать опухоли при перевивке животным) ведет себя как признак рецессивный — гетерокарионы, образованные слиянием нормальных и трансформированных (опухолевых) клеток, ведут себя как нормальные. Таким образом, в нормальных клетках явно присутствуют факторы, тормозящие клеточное деление и способные при внесении в опухолевую клетку нормализовать ее. Многие из этих белковых факторов идентифицированы; кодирующие их гены получили название генов-супрессоров или антионкогенов. Итак, полная опухолевая трансформация клетки является следствием нескольких генетических событий — активации онкогена(ов) и инактивации гена(ов), осуществляющих супрессорные функции.
Рис. 3. Схематическое представление клеточного цикла (пояснения в тексте).
Клетки организма находятся в одном из трех возможных состояний (рис. З): 1 — в митотическом цикле, 2 — в стадии покоя с сохранением возможности вернуться в цикл и, наконец, 3 — в стадии окончательной дифференцировки, при которой способность делиться полностью утрачена (таковы, например, нейроны головного мозга). Образовывать опухоли могут, естественно, только клетки, способные делиться. Цикл удвоения разных клеток человека существенно варьирует: от 18ч у клеток костного мозга до 450ч. у клеток крипт толстой кишки. Основными его периодами являются митоз (М) и синтез ДНК (фаза S), между которыми выделяют два промежуточных периода — G1 и G2. Во время интерфазы (период между двумя делениями) клетка растет и готовится к митозу. На протяжении фазы G1 существует ответственный момент (т.н. точка рестрикции R), когда решается, войдет ли клетка в следующий цикл деления или предпочтет стадию покоя G0, в которой она может находиться неопределенно долго до получения митогенного сигнала. При делении клетки также имеются два критически важных момента: фаза синтеза ДНК и вхождение в митоз, когда действуют своеобразные «контрольно-пропускные пункты» (checkpoints). В этих пунктах проверяется готовность к удвоению (репликации) ДНК — в первом случае и завершенность репликации — во втором. Любое стрессорное воздействие (например, отсутствие питательных веществ, гипоксия и, в особенности, повреждение ДНК) блокирует движение клетки по циклу в одном из двух контрольных пунктов (checkpoints). Следовательно, блокируется этап способный закрепить повреждения ДНК и передать их потомству. Аналогичная цель достигается апоптозом, причем какой путь выберет клетка (блок деления или апоптоз) зависит от многих условий. Если же дефектны сами «контрольно-пропускные пункты», то дефекты генома не устраняются, передаются потомству и возникает опасность злокачественной трансформации клетки. В механизме checkpoint участвуют комплексы белков. Их совокупность образуют систему «тормозов», не позволяющих клетке делиться в отсутствие адекватных стимулов и при наличии повреждений ДНК. Кодирующие эти белки гены получили название генов-супрессоров. Особое значение этой системы заключается в том, что опухолевая трансформация клетки возможна только после инактивации генов-супрессоров. Важно при этом иметь в виду, что в соматической клетке существуют по два аллеля каждого из генов, в том числе и генов-супрессоров, и, следовательно, для их инактивации необходимы два независимых события (например, делеция одного аллеля и мутация другого). Именно в этом кроется причина того, что «спорадические» опухоли возникают относительно редко (вероятность возникновения в одной клетке нескольких независимых мутаций, причем поражающих один и тот же локус обеих хромосом, относительно невелика), а «семейные» чрезвычайно часты (в «раковых» семействах один из двух наследуемых аллелей того или иного гена-супрессора исходно дефектен). В последнем случае система «тормозов» у всех клеток данного организма держится лишь на одном нормальном аллеле, что резко снижает ее надежность и повышает риск возникновения опухоли. Именно это и происходит при наследственной ретинобластоме и других наследственных заболеваниях. Главным клеточным «хранителем генома» является белок-супрессор р53. Белок р53 активируется при возникновении различных повреждений ДНК и, в частности, при гамма-облучении клеток. В клетках с интактной ДНК содержание р53 исчезающе мало и соответственно ничто не препятствует активации группы генов, необходимых для вступления клетки в фазу синтеза ДНК. У клеток с дефектным или отсутствующим р53 контрольный пункт G1/ S неполноценен. Это проявляется в том, что повреждения ДНК, индуцированные ионизирующей радиацией или каким-либо другим способом, не приводят ни к задержке клеток на границе фаз G1/S, ни к апоптозу. В результате в популяции накапливаются клетки со множественными нарушениями структуры ДНК; появляется и со временем нарастает нестабильность генома, которая ведет к появлению все новых клеточных клонов. Их естественный отбор лежит в основе опухолевой прогрессии — постоянного «дрейфа» опухоли ко все большей автономности и злокачественности. Именно свойство нестабильности генома наделяет клетки опухоли, с одной стороны, замечательной приспособляемостью к условиям окружающей среды и «увертливостью» по отношению к лечебным воздействиям, а с другой — все новыми качествами, в частности, способностью метастазирования.
3. Стадии развития опухоли Канцерогенез является многоступенчатым процессом. Лучше всего это было продемонстрировано при экспериментальном изучении химического канцерогенеза. Выделяют три основные стадии химического канцерогенеза — инициация, промоция и прогрессия. Инициация — начальный этап в длинной цепи событий, ведущих к образованию опухолевого очага. Оно заключается в мутации одного из генов, регулирующих клеточное размножение. Клетка становится «инициированной», т.е. потенциально способной к неограниченному делению, но требующей для проявления этой способности ряда дополнительных условий. Инициирующими факторами являются различные канцерогены, вызывающие повреждение ДНК. Промоция. К промоторам относятся химические вещества, которые не вызывают повреждения ДНК, т.е. не являются канцерогенами, но при длительном воздействии на инициированные клетки способствуют возникновению опухоли. По-видимому, промоторы стимулируют клеточное деление, что создает критическую массу инициированных клеток и способствует их высвобождению из-под тканевого контроля. Лучше всего изучен промотор – форболовый эфир, мощный активатор протеинкиназы С, фосфорилирующей ряд субстратов сигнальной трансдукции. Форболовый эфир вызывает также продукцию фактора роста некоторыми клетками. Его способность активировать протеинкиназу С опирается на структурное сходства этого эфира с 1,2 диацилглицерином - физиологическим активатором протеинказы С. Другие промоторы воздействуют на сигнальную трансдукцию с помощью родственного, но слабого механизма. Итак, если воздействие инициатора может вызвать мутационную активацию онкогена, например гена ras, то последующий эффект промоторов приводит к пролиферации и клональной экспансии инициированных клеток – мутантов. При пролиферации клеток возрастает риск мутаций и опухолевой трансформации. Таким образом, главное звено промоции – размножение инициированных клеток – существенно облегчает завершение процесса злокачественной трансформации. Прогрессия. Опухоль является потомством, т.е. клоном, одной первичной клетки, которая в результате многостадийного процесса приобрела способность нерегулируемого роста. Распространение опухоли по всему организму (метастазирование) также является следствием размножения клона первично трансформированной клетки. Однако, геном опухолевой клетки нестабилен, поэтому в процессе многократного деления появляются новые клоны, различающиеся генотипически и фенотипически. Это свойство определяет прогрессию опухоли, т.е. появление в результате естественного отбора клонов, обладающих быстрым ростом и устойчивостью к факторам защиты организма. Опухоль становится все более агрессивной, автономной и устойчивой к лечебным воздействиям. Таким образом, непрерывно прогрессируя, претерпевая качественные изменения и проходя через необратимые стадии, опухоль «дрейфует» в направлении увеличения злокачественности. Т.о. рост опухоли — это не только увеличение числа однородных клеток. Опухоль постоянно претерпевает качественные изменения и приобретает новые свойства — все большую автономность от регулирующих воздействий организма, деструктивный рост, инвазивность, способность к образованию метастазов (обычно отсутствующую на ранних этапах) и, наконец, ее поразительную приспособляемость к меняющимся условиям. Признаки злокачественности возникают в ходе прогрессии независимо друг от друга, что объясняет разнообразие опухолевых фенотипов в отличие от нормально развивающейся ткани, структура и функция которой всегда четко определены.
IV. Биологические особенности опухолевого роста. Биологические особенности опухолевого роста проявляются атипизмом, то есть ненормальностью свойств опухолевой клетки и ткани. Можно выделить следующие биологические особенности опухолевого роста: Атипизм обмена веществ Атипизм структуры опухолевой клетки и ткани Функциональный атипизм Атипизм регуляции опухолевых клеток Атипизм размножения и развития
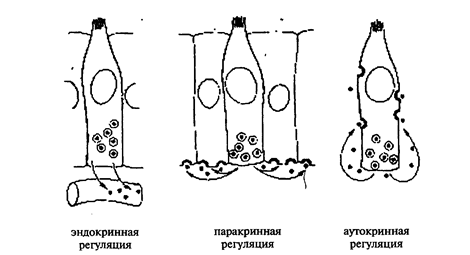
1). Атипизм энергетического и углеводного обмена проявляется усилением гликолиза и ослаблением тканевого дыхания. В опухолевой ткани усилены также пентозо-фосфатный шунт и использование в нем глюкозы, что приводит к повышенному образованию рибозы и НАДФН2, которые необходимы для синтеза нуклеиновых кислот и размножения клеток. Указанные изменения обусловлены: уменьшением васкуляризации и оксигенации опухолевой ткани вследствие редукции сети капилляров. возрастанием активности гликолитических ферментов и угенетением ферментов тканевого дыхания. изменением изоферментного спектра, что позволяет опухолевой клетке конкурировать с нормальной за субстрат (в частности за глюкозу). Опухоль становится «ловушкой» для глюкозы и приобретает устойчивость к гипоксии в условиях инвазивного роста и метастазирования В опухолевой ткани возникает лактацидоз, который может отрицательно действовать на окружающие и отдаленные ткани. 2). Атипизм жирового обмена проявляется преобладанием липогенеза над липолизом с образованием липидов, идущих на построение мембран вновь образующихся клеток, в том числе холестерина и фосфолипидов. 3). Атипизм белкового обмена проявляется усилением синтеза белков с активным поглощением аминокислот из крови даже при низкой их концентрации («ловушка азота») и изменением качественного состава белков опухолевой клетки. Это в свою очередь проявляется (помимо синтеза онкобелков): -изменением изоферментного спектра опухолевых клеток, -прекращением синтеза органоспецифичных белков (альбуминов в печени), -синтезом эмбриональных белков (адьфа-фетопротеин при первичном раке печени), -синтезом гетероорганных белков и полипептидов (АКТГ, паратгормон при бронхогенном раке легких), -синтезом аномальных белков (аномальные иммуноглобулины при болезни тяжелых цепей Франклина, макроглобулинемии Вандельстрема, белок Бенс-Джонса при миеломной болезни). Атипизм белкового обмена приводит не только к безудержной пролиферации опухолевых клеток, но и ускользанию их от иммунного надзора, т.к. снижение синтеза органоспецифических белков ведет к антигенному упрощению опухолевых клеток, а эмбриональные белки имеют низкую антигенную активность. Организм в целом лишается незаменимых аминокислот, а появление аномальных белков, в том числе гормонов, становится причиной изменения состава крови и микроциркуляции, повреждения неопухолевых органов. В опухолевых клетках также усилен синтез нуклеиновых кислот. 4). Атипизм водно-минерального обмена проявляется увеличением в опухолевой ткани внутриклеточного К+ и снижением Са2+, (при этом в самом организме развивается гиперкальциемия) что ведет к нарушению межклеточных связей, отшнуровке клеток от опухолевого узла, инвазивному росту и метастазированию. Этому также способствует повышение величины«–«заряда на поверхности опухолевых клеток и их взаимное отталкивание. Для опухолевой ткани характерна гипергидратация из-за гипоонкии крови. 2. Атипизм структуры опухолевой ткани – вопрос подробно рассматривается в курсе патологической анатомии. 3. Функциональный атипизм может проявляться: 1) – снижением или утратой специфических функций (миелобласты неспособны к фагоцитозу, опухолевые клетки печени не синтезируют альбумин, снижается секреция желудочного сока при аденокарциноме желудка). 2) - активацией функций (увеличение выработки ИЛ-2 при Т-клеточном лейкозе человека, гормон продуцирующие опухоли). 3) - извращением функций – опухолевые клетки начинают выполнять несвойственные им функции (выработка АКТГ эпителием бронхов при раке легких, кальцитонина – при раке молочной железы). 4. Атипизм регуляции опухолевых клеток заключается в следующем: 1) Автономность – т.е. независимость опухолевой клетки от тормозных командных влияний различных систем организма – нервной, эндокринной, иммунной, и от тормозных влияний, связанных с межклеточным взаимодействием. Но эта автономность носит относительный характер, т.к. регулирующие влияния, способствующие реализации генетической программы опухолевой клетки воспринимаются в полной мере. Так, опухолевая клетка не реагирует на тормозящие пролиферацию влияния кортикостероидов, но обладает повышенной чувствительностью к инсулину. 2). Аутокринная регуляция – опухолевая клетка сама продуцирует факторы роста, рецепторы к ним и их аналоги, т.е. стимулирует свое собственное размножение. 3). Паракринная регуляция – под влиянием команд опухолевой клетки окружающие ткани вместо выработки факторов, тормозящих пролиферацию (механизм контактного торможения в норме) начинают вырабатывать факторы - стимуляторы пролиферации.
Рис. 4. Схематическое представление эндокринной, паракринной и аутокринной регуляции.
5. Атипизм размножения и развития характеризуется следующим: 1). Иммортализация – «бессмертие» клеток, т.е. опухолевые клетки преодолевают «барьер Хейфлика» (запрограммированное число делений для каждого вида клеток) и начинают размножаться неопределенно долго (клетки карциномы Эрлиха перевивают с 1905 года). Нормальная соматическая клетка при каждом делении теряет часть ДНК на концевом участке хромосомы – теломере, т.к. ДНК-полимераза не может начинать синтез с первого нуклеотида. Потеря концевой ДНК делает невозможной бесконечную пролиферацию, что объясняет барьер Хайфлика. Иммортальные клетки, в том числе опухолевые, содержат фермент теломеразу, достраивающий хромосому. 2). Увеличивается число делящихся клеток (30-50%, даже 100%), в некоторых опухолевых клетках увеличивается скорость митотического цикла, что ведет к гиперплазии. 3). Анаплазия – изменения биологических свойств опухолевых клеток, которое делает ее похожей на эмбриональную или недифференцированную клетку. В опухолевой клетке и ткани имеет место блок созревания и дифференцировки, причем, чем он выше, ближе к стволовой клетке, тем более остро и злокачественно протекает опухолевый процесс. Анаплазия также проявляется всеми видами атипизма указанными выше. Отличие злокачественных опухолей от доброкачественных и клинико морфологические стадии опухолевого роста – рассматривались в курсе патологической анатомии.
|