
Механизмы реакций, протекающих на поверхности кислотных катализаторов.
Внутримолекулярная и межмолекулярная дегидратация. Кислотные катализаторы ускоряют как внутри-, так и межмолекулярную дегидратацию гидроксилсодержащих углеводородов. Активность и селективность катализаторов (помимо условий проведения процесса) зависит от кислотности поверхности. Рассмотрим пример каталитических превращений этанола на различных образцах промышленного Al2O3, различающихся содержанием технических примесей - SiO2 и Na2O (Табл. 5.22).
Таблица 5.22. Поведение оксида алюминия в дегидратации этанола.
* - ммоль NH3, адсорбируемого 1 граммом Al2O3.
Проанализируем данные Таблицы 5.22. Увеличение содержания SiO2 приводит к усилению Бренстедовской кислотности. Увеличение содержания Na2O приводит нейтрализации сильных Бренстедовских кислотных центров, и к стабилизации основных центров. Очевидно, что общая кислотность образцов растет с увеличением содержания SiO2 и уменьшением содержания Na2O. Установлено, что дегидратация этанола протекает при согласованном действии кислотных протонных и основных каталитических центров:
Поэтому наивысшая активность и селективность по этилену наблюдается на образце со средней кислотностью (оптимальное соотношение основных и кислотных центров). Образец с наименьшей кислотностью обладает самой низкой активностью (конверсия) и наименьшей селективностью по этилену. Кроме того было установлено, что дегидратация протекает на кислотных центрах Бренстеда средней силы, а Льюисовские центры практически не катализируют эту реакцию. Этот вывод был сделан на основании того факта, что добавка небольших количеств оснований Льюиса (аммиак, пиридин и т.п.), блокирующих в первую очередь сильные кислотные и апротонные центры, не ингибируют реакцию. Образование эфира на поверхности Al2O3 протекает по механизму Лэнгмюра-Хиншельвуда, при котором взаимодействие происходит между алкоголятом (С2Н5О-), образовавшемся на основном центре и молекулой этанола, активированной водородной связью на соседнем кислотном центре. Поэтому, чем больше содержание Na2O, тем выше селективность образования эфира. Важный момент в данном примере - образование кокса. Как видно количество кокса растет пропорционально росту Бренстедовской кислотности. Это связано с тем, что именно на этих центрах образуются промежуточные карбокатионы, инициирующие димеризацию и олигомеризацию этилена. Отложение нелетучих продуктов олигомеризации приводит к блокированию поверхности, снижению каталитической активности и коксообразованию.
Реакции в каталитическом крекинге и реформинге (крекинг, изомеризация, полимеризация, циклизация, ароматизация, коксообразование)
Ключевой стадией, обеспечивающей каталитическую активность кислотных катализаторов в реакциях крекинга, изомеризация, полимеризация, циклизация, ароматизация, коксообразования, является образование карбокатионов при взаимодействии углеводородов с активными центрами поверхности. Образование карбокатиона может протекать по трем механизмам: - Отрыв гидрид-аниона от парафина сильными кислотными центрами Льюиса:
- Протонирование парафина кислотными центрами Бренстеда с образованием водорода и карбокатиона. Такая реакция возможна только при участии очень сильных Бренстедовских центров, например, расположенных рядом с Льюисовским центром ((5.59), (5.64)):
- Протонирование олефина:
Н+ + R1-CH=CH-R2 ßà R1-CH2-+CH-R2 (5.69)
Олефины, необходимые для протекания реакции (5.69) могут присутствовать в сырье как примесь, образовывать в результате термического крекинга или в результате крекинга карбокатионов, образующихся на каталитических центрах по реакциям (5.67), (5.68). Крекинг неразветвленного вторичного карбокатиона протекает по схеме:
R1-CH2-CH2-+CH-CH2-R2 ßà R1-CH=CH2 + +CH2-CH-R2 (5.70)
Образовавшийся первичный карбокатион может перегруппироваться в более устойчивый вторичный или третичный. Один из возможных механизмов циклизации и ароматизации углеводородов на кислотных катализаторах может быть представлен следующей схемой. Олефины способны отдавать аллильный гидрид-анион кислотным центрам катализатора с образованием резонансно-стабилизированного аллильного крбокатиона:
(5.71)
Образовавшийся карбокатион может отдавать протон основному центру каталитической поверхности с образованием диена:
Далее по такому же механизму может образоваться триен, который легко циклизуется при катализе протонными кислотными центрами:
Образовавшийся циклический диен при превращается в карбокатион при взаимодействии с другим карбокатионом на поверхности катализатора:
Далее, отдавая протон основному центру катализатора, крбокатион превращается в ароматический углеводород:
Циклизация может протекать и в результате внутримолекулярной электрофильной атаки в карбокатионе, имеющем двойную связь:
Как видно из приведенных схем ароматизация углеводородов на кислотных катализаторах протекает с участием основных центров. Но так активность основных центров в кислотных катализаторах низка, то реакции ароматизации протекают медленно. При каталитическом крекинге газойля доля ароматических углеводородов в продуктах составляет порядка 2%. Кислотно-каталитические реакции изомеризации, полимеризации, алкилирования, переалкилирования (диспропорционирования) на гетерогенных катализаторах протекают по тем же механизмам, что и при катализе гомогенными кислотами. Образование кокса на поверхности кислотных катализаторов связано главным образом с кислотно-каталитической реакцией конденсации ароматических соединений. Упрощенная схема может быть представлена так:
Многоядерный ароматический карбокатион очень устойчив, поэтому он может не только отдавать протон с образованием полиядерного соединения:
но и продолжать цепочку реакций конденсации дальше с образованием кокса. Эта упрощенная схема образования кокса может сопровождаться реакциями алкилирования, полимеризации, циклизации.
|
 (5.66)
(5.66)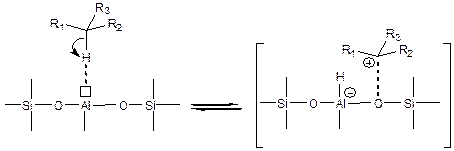 (5.67)
(5.67) (5.68)
(5.68)
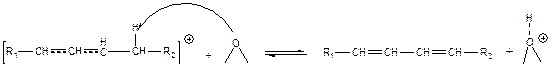 (5.72)
(5.72)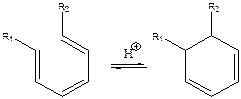 (5.73)
(5.73) (5.74)
(5.74)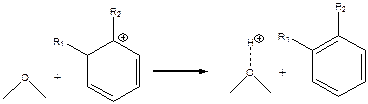 (5.75)
(5.75) (5.76)
(5.76) (5.77)
(5.77) (5.78)
(5.78)


