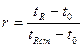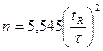КАК ВЫБИРАЮТ УСЛОВИЯ РАБОТЫ ХРОМАТОГРАФИЧЕСКОЙ КОЛОНКИ
Однако вернемся немного назад. Мы ввели пробу в колонку. Что происходит с этой пробой? Она поступила на начальный участок колонки, где, как и в других частях колонки, есть зерна, пропитанные нелетучей жидкостью, а между зернами находится газ. «Остановим мгновение» и посмотрим, как будет вести себя анализируемое вещество. Часть его будет оставаться в газе между зернами, а другая часть растворится в жидкости. Другими словами, произойдет распределение сорбата между газом и жидкостью. Но в какой пропорции? Какая часть останется в газе, а какая растворится? Эта пропорция есть коэффициент распределения Г, который равен отношению концентрации анализируемого сорбата в жидкости сж (число молей сорбата в I см3 жидкости) к концентрации его в газе сг, (число молей сорбата в 1 см3 газа). Таким образом, мы можем записать: Выяснив, что произошло сразу после ввода пробы в колонку, рассмотрим нашу систему в движении. Газ движется между зернами. Поэтому после того как в первый момент в начале колонки сорбатраспределится между газовой и жидкой фазами, газ сразу же «унесет» свою часть дальше, в глубь колонки, а свежая порция газа, занявшая это место, «потребует» у неподвижной жидкости своей доли с тем, чтобы распределение сорбата междуфазами снова соответствовало коэффициенту Г. С другой стороны, газ, который транспортирует сорбат, соприкоснется в глубине колонки со свежими порциями неподвижной жидкости и вынужден будет отдать ей львиную долю своей добычи, так как коэффициент Г гораздо больше единицы. Таким образом, молекуле сорбата не остается ничего иного, как «нырять» из одной фазы в другую, пока, наконец, газ не вынесет ее прочь из колонки. Другими словами, хроматографический процесс есть цепь сорбционных и десорбционных актов, причем под сорбцией, как мы уже говорили, понимают либо растворение в неподвижной жидкости, либо адсорбцию на поверхности адсорбента, если он служит в колонке неподвижной фазой. Десорбция же — это обратный процесс перехода вещества в газовую фазу. Газ при своем движении очищает колонку от сорбата, вымывает, элюирует его или проявляет хроматограмму. С какой же скоростью передвигаются молекулы сорбата вдоль колонки? Очевидно, что эта скорость зависит и от скорости газа-носителя, и от коэффициента распределения Г. Действительно, чем быстрее движется газ, тем на большее расстояние он успевает перенести молекулу сорбата. С другой стороны, чем больше время пребывания молекулы сорбата в жидкости (т.е. чем больше Г), тем больше она «отстает» от движущегося газа. Кроме того, безусловно, что чем больше в колонке жидкости, тем больше время пребывания молекул сорбата в ней. Обозначим линейную скорость газа-носителя через а (см/с), а скорость движения зоны сорбата через и (разумеется, здесь мы имеем в виду лишь скорость перемещения вдоль колонки, так как в действительности молекула сорбата «успевает» и переходить из одной фазы в другую, и перемещаться в различных направлениях, как в газе, так и в жидкости). Тогда
где tг— время пребывания молекул сорбата в газовой фазе, а tж — время пребывания ее в жидкости. Если бы объем жидкой фазы в колонке был равен объему газовой, то было бы справедливо равенство
Но газовая фаза занимает в колонке больший объем (wг) по сравнению с объемом, занимаемым жидкой фазой (wж). Поэтому следует записать:
Из последнего соотношения можно сделать очень важный вывод о том, что скорость, с которой сорбат движется вдоль колонки это специфическая характеристика . Если проба содержит не один, а два сорбата, у которых различны значения Г, то они будут передвигаться по колонке с разными скоростями. Таким образом, вещества с разными Г будут разделяться в хроматографической колонке.
Задача исследователя, который желает разделить смесь на составляющие, как раз и должна заключаться в том, чтобы подобрать такую неподвижную жидкость, которая по-разному растворяла бы составляющие смеси. Конечно, задача эта тем сложнее, чем больше компонентов входит в состав смеси. Рассматривая законы движения веществ по колонке, нужно учесть одно важное обстоятельство. Мы говорили о том, что коэффициент распределения данного вещества между газом и жидкостью (Г) — величина постоянная. Это значит, что какова бы ни была проба, большая или малая, какова бы ни была концентрация вещества в газе (сг) все равно распределение между фазами произойдет в одной и той же пропорции. На графике сж—с, изотерма сорбции при постоянном Г будет иметь вид прямой линии. В этом случае вся зона анализируемого сорбата, все части этой зоны будут передвигаться с практически одинаковой скоростью и перо самописца запишет узкий симметричный пик (подобный нарисованному на графике около прямой б). Но такой случай идеальный. На практике, особенно при использовании в колонке адсорбента, коэффициент Г зависит от концентрации вещества (сорбата); и изотермы сорбции принимают вид выпуклой (а) или вогнутой (в) кривой. Это говорит о том что разные участки зоны сорбата будут передвигаться с разными скоростями: в одних случаях какая-то зона отстает, образуется «хвост»— мы говорим «происходит размытие тыла» (кривая а), в других быстрее выходит начальная зона, как бы забегая вперед, и мы говорим «размытие фронта» (кривая в). При этом происходит наложение «хвоста» одного компонента смеси на «голову» другого, ухудшается разделение смеси. Хроматографические пики получаются несимметричными: в первом случае более полога конечная ветвь, во втором — начальная. Но если такое неприятное явление устранить, то каждое вещество будет передвигаться по слою сорбента со своей постоянной скоростью и выходить из колонки в строго определенное время tR. Эту величину называют в хроматографии временем удерживания; она зависит от длины колонки L и силы сорбции:
А нельзя ли по времени удерживания «узнавать» вещество? Оказывается, вполне можно, и именно на этом принципе основан качественный хроматографический анализ, идентификация («узнавание») компонентов анализируемых смесей. Однако для получения сравнимых данных исследователи должны были проводить эксперименты в строго одинаковых условиях —на колонках одной длины, при одной и той же скорости газа и при одинаковом количестве неподвижной жидкости. Но это практически осуществить невозможно. Поэтому лучше использовать так называемое относительное удерживание г, то есть разделить время удерживания анализируемого вещества на время удерживания какого-нибудь другого вещества, которое мы будем использовать в качестве стандарт ого. Чтобы относительное удерживание зависело только от неподвижной фазы, нужно ввести поправку на газовое пространство в колонке. Введя все эти поправки, получают следующую формулу для расчета:
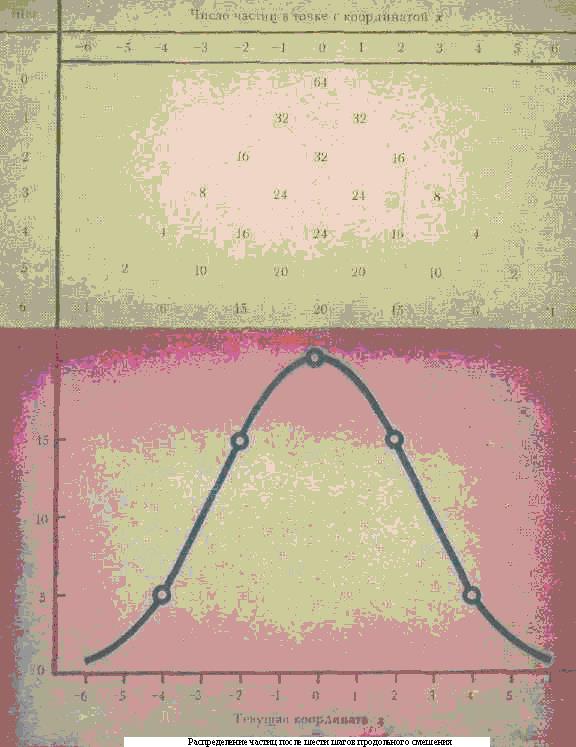
Таким образом, чтобы рассчитать г, нужно знать время удерживания анализируемого вещества tRстандарта tRcт и какого-нибудь газа, который не сорбируется в колонке, t0(гелия, иногда воздуха). В литературе по газовой хроматографии приводят таблицы, которые содержат большое число данных по относительному удерживанию самых разнообразных веществ на колонках с разными сорбентами. Однако при проведении хроматографического процесса нужно учитывать и еще одно обстоятельство. Разделить вещества было бы не так уж сложно, если бы каждое из них выходило из колонки (элюировалось) компактно. Но хроматограмме ему соответствовал бы узкий пик, высота которого говорила о количестве выделенного вещества. Но на практике это не так, даже если коэффициент распределения Г постоянен и асимметричного размытия зон нет. Во-первых, молекуле сорбата требуется определенное время, чтобы, будучи в газовой фазе, пройти расстояние до поверхности неподвижной жидкости, раствориться в ней, пройти определенный путь внутри жидкости (продиффундиро-вать) и освободиться. Естественно, что за это время те молекулы, сорбата, которые находились в движущейся газовой среде, успели пройти определенный путь вдоль колонки (и чем больше скорость газа-носителя, тем этот путь больше). Таким образом, происходит расширение (размытие) хроматографической зоны. Эти причины размытия зон называют кинетическими факторами. Во-вторых, нужно учитывать хаотическое движение молекул в жидкой и особенно в газовой фазах. Этот процесс сопровождался продольным «расплыванием» хроматографической зоны. Попробуем чисто умозрительно представить себе, как это происходит. Пусть вначале на прямой линии в точке 0 находилось 64 частицы. Вследствие хаотического движения частицы с равной вероятностью смещаются вправо и влево, причем через промежуток времени, необходимый для первого шага, очевидно, справа (точка +1) и слева (точка -1) будут находиться по 32 частицы. Следующим шагом будет смещение частиц, которые оказались в каждой из образовавшихся групп, вправо и влево и т д. Нижняя шестая строка показывает распределение частиц после шестого шага. Если это распределение изобразить графически, то получится симметричная кривая. При достаточно большом числе шагов смещения, что имеет место в действительности, кривая, характеризующая форму хроматографической зоны, принимает вид известной в физике и математической статистике колоколообразной кривой распределения ошибок гауссовой кривой). Разумеется,, чем больше скорость газа-носителя, тем меньше время пребывания зоны сорбата и, следовательно, тем меньше размытие, вызванное продольной диффузией. Но, к сожалению, очень высокой скорость делать нельзя, так как зона будет расширяться из-за влияния кинетических факторов. Есть и другие причины размытия зон при высоких скоростях: это, например, известный в гидравлике «стеночный эффект». В газовой хроматографии оптимальной считают линейную скорость газа-носителя 5—10 см/с, именно при этих условиях удается получать наиболее узкие зоны сорбатов. Теперь мы можем сказать, что разделение веществ в хроматографической колонке определяется двумя основными факторами: во-первых, селективностью неподвижной фазы, т.е. ее свойством по-разному сорбировать компоненты смеси, и, во-вторых, эффективностью колонки, т. е. способностью давать узкие хроматографические пики. Если подобрана селективная неподвижная фаза, но эффективность колонки была низкой, то в результате хроматографического разделения произойдет перекрывание зон (А). Если эффективность колонки достаточно высока, то неподвижная жидкая фаза практически одинаково растворяет компоненты смеси (коэффициенты Г для обоих веществ равны), хроматографический пик будет узким, но смесь разделить также не удастся. Оба вещества выйдут из колонки вместе (Б). Только если подобрать селективную жидкую фазу и колонку высокой эффективности, разделение зон будет хорошим (В). Как подобрать селективную жидкую фазу, можно себе представить, если вспомнить, что это значит. Просто такая жидкость должна по-разному растворять компоненты смеси, т. е. коэффициенты Г компонентов смеси для данного растворителя должны быть разными, и чем больше они будут различаться, тем лучше. А как оценить эффективность хроматографической колонки? По аналогии с ректификационной колонкой эффективность хроматографической колонки стали оценивать числом теоретических тарелок. В хроматографии эта величина определяется отношением времени удерживания вещества к ширине пика, т. е. скоростью размытия зоны. Чем меньше скорость размытия, тем эффективнее колонка. Причем эта зависимость не простая. Так, если ширину пика, измеренную на половине его высоты, обозначить через х., то число теоретических тарелок п будет
Сравнивая хроматографические колонки с ректификационными, мы видим, что последние по своей эффективности остаются далеко позади. Самая обычная метровая колонка в хроматографии имеет эффективность в сотни теоретических тарелок, в то время как эффективность ректификационной колонки такой же длины на целый порядок меньше. Это и позволяет хроматографически разделять вещества, у которых значения Г различаются очень не намного, например близкокипящие изомеры или молекулы, содержащие изотопы, и т.д.
Степень разделения К равна разности времен удерживания, деленной на сумму ширин соседних пиков: К = и, как видно из приведенной формулы, зависит от числа теоретических тарелок. Чем длиннее колонка, тем выше ее эффективность. Зная значения входящих в формулу величин, можно определить, колонка какой длины нужна для разделения той или иной пары веществ. При этом следует помнить, что оптимальными являются условия, когда К близка к единице. Ну, а если, несмотря ни на что, вещества не разделяются? Значит, величины коэффициентов распределения у них одинаковы и, сколько ни удлиняй колонку, эффекта не будет. Тогда нужно взять другую неподвижную фазу, т.е. изменить селективность колонки, В этом случае дробь в правой части уравнения, которая содержит времена удерживания сорбатов (коэффициент селективности) изменится. Кстати, в ректификации этого сделать нельзя, так как степень разделения там определяется только температурами кипения разделяемых веществ. В газовой хроматографии в качестве неподвижной фазы используют сотни разных жидкостей: это и неполярные парафины, и полярные соединения типа гликолей и их эфиров, и вещества, содержащие цианэтильные группы, и т, д. Широко применяют эфиры фталевой кислоты *, которые известны в промышленности как пластификаторы при производстве пластмасс. Казалось бы, всегда можно найти то, что годится для разделения анализируемой смеси. Но бывают и довольно курьезные случаи. Так, оказалась очень трудной задача разделения смеси этилбензола и трех изомерных ксилолов. На колонке с обычными неподвижными фазами удается получить три пика: этилбензол и ор- то-ксилол выходят отдельно, а мета-ксилол и «ара-ксилол вместе. Но вот нашли, что органическое производное» глины — бентон хорошо разделяет мета- и «ара-изомеры. Казалось бы, задача решена. Однако полностью разделить смесь всех четырех веществ снова не удалось: бентон оказался настолько «хорош», что «ара-ксилол, обгоняя.мета-ксилол, догонял этилбензол, а.мета-ксилол отставал и выходил из колонки вместе с орто-ксило-лом. Глядя на такие хроматограммы, можно решить только одно: надо «испортить» бентон, сделать его менее селективным при разделении мета- и пара-изомеров, и тогда все четыре компонента будут выходить через почти равные промежутки времени. Для этого попытались смешать бентон с какой-нибудь «плохой» фазой, например с обычным вазелиновым маслом, т.е. использовать смешанную неподвижную фазу. В этом случае на хроматограмме получились четыре пика, т.е. смесь удалось разделить. Для оценки оптимальности подобранных условий разделения смеси в хроматографии ввели особую величину — коэффициент равномерности. Если сравнивать его значения для самых разных * Эти соединения многим известны, особенно туристам, потому что ими пользуются для отпугивания насекомых Скорость анализа с учетом степени разделения компонентов оценивают с помощью коэффициента быстродействия, который соответствует числу хорошо(сК — не менее единицы) разделенных компонентов, выходящих из колонки за одну минуту:
Здесь nк— число пиков на хроматограмме, t — время анализа, а степень разделения К относится к наихудшим образом разделяемой паре компонентов (пиков на хроматограмме). .
Быстрота разделения — это одно из важных преимуществ газовой хроматографии перед другими методами. Обычные лабораторные анализы проводят за 20— 30 минут, а разделение смесей, включающих десятки компонентов — за час или даже несколько часов. На промышленных же предприятиях, где самое небольшое промедление может сильно повредить качеству продукта, результаты анализа должны выдаваться за считанные минуты. Вот здесь-то особенно важно использовать хроматографические методы. Есть такие из них, которые позволяют разделять смесь 5—10 компонентов за несколько секунд!
|
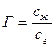 Коэффициент Г-это постоянная величина для данного сорбата и данной жидкости, она зависит только от температуры. Постоянство коэффициента Г означает, что в каждое мгновение существует равновесие между газом и жидкостью. Но такое равновесие совсем не означает, что какая-то молекула пробы будет все время находиться в жидкости, а другая — все время в газе между зернами. Нет, равновесие — это процесс динамический, каждая молекула какую-то часть времени проводит в жидкости и какую-то — в газе. А отношение этих очень малых промежутков времени также равно Г.
Коэффициент Г-это постоянная величина для данного сорбата и данной жидкости, она зависит только от температуры. Постоянство коэффициента Г означает, что в каждое мгновение существует равновесие между газом и жидкостью. Но такое равновесие совсем не означает, что какая-то молекула пробы будет все время находиться в жидкости, а другая — все время в газе между зернами. Нет, равновесие — это процесс динамический, каждая молекула какую-то часть времени проводит в жидкости и какую-то — в газе. А отношение этих очень малых промежутков времени также равно Г.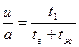
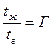
 и тогда
и тогда