Требования, предъявляемые к лекарственным средствам
1) требования к качеству и безопасности лекарственных средств; 2) требования к упаковке и маркировке лекарственных средств. Требования к маркировке и упаковке лекарственных средств. 1. Упаковка лекарственных средств должна обеспечивать их безопасность и неизменность ее идентификационных признаков при обращении лекарственных средств в течение срока годности. 2. Лекарственные препараты, за исключением изготавливаемых в аптечных организациях, поступают в обращение, если на первичной и вторичной (потребительской) упаковках хорошо читаемым шрифтом на русском языке указаны: На первичной упаковке: 1) торговое наименование лекарственного средства и международное непатентованное наименование; 3) дозировка и количество в первичной упаковке; 3) номер серии; 4) срок годности; На вторичной (потребительской) упаковке: 1) торговое наименование лекарственного средства и международное непатентованное наименование; 2) название организации - производителя лекарственных средств; 3) номер серии; 4) способ применения; 5) дозировка и количество доз в упаковке; 6) срок годности; 7) условия отпуска; 8) условия хранения; 9) меры предосторожности при применении лекарственных средств. 3. Фармацевтические субстанции поступают в обращение, если на вторичной (потребительской) упаковке хорошо читаемым шрифтом на русском языке указаны: 1) торговое наименование лекарственного средства и международное непатентованное наименование; 2) название организации - производителя лекарственных средств; 3) номер серии и дата изготовления; 4) количество в упаковке и единицы измерения количества; 5) срок годности; 6) условия хранения. 4. Сыворотки поступают в обращение с указанием, из крови, плазмы крови, органов, тканей какого животного они получены; вакцины - с указанием питательной среды, использованной для размножения вирусов и бактерий. 5. Лекарственные средства, зарегистрированные как гомеопатические, имеют надпись: «Гомеопатические». 6. Лекарственные средства, предназначенные для лечения животных, имеют надпись: «Для животных». 7. Лекарственные средства, полученные из растительного сырья, имеют надпись: «Продукция прошла радиационный контроль». 8. Лекарственные средства, предназначенные для клинических исследований, имеют надпись: «Для клинических исследований». 9. Лекарственные средства, предназначенные исключительно для экспорта, маркируются в соответствии с требованиями страны-импортера. 10. Лекарственные препараты должны поступать в обращение только с инструкцией по применению лекарственного препарата, содержащей следующие данные на русском языке: · торговое наименование, международное непатентованное, химическое или иное наименование лекарственного средства; · лекарственная форма с указанием количественного содержания (активности) действующих веществ и наименований вспомогательных веществ; · фармакотерапевтическая группа лекарственного препарата; · название, юридический и фактический адреса организации-производителя лекарственного препарата; · показания к медицинскому применению; · противопоказания к медицинскому применению; · режим дозирования и путь введения, длительность лечения, если её следует ограничить; · возможные побочные действия, нежелательные явления и серьёзные нежелательные явления при медицинском применении лекарственного препарата; · взаимодействие с другими лекарственными препаратами и (или) пищевыми продуктами; · указание о возможности медицинского применения у беременных, женщин, кормящих грудью, несовершеннолетних; · срок годности, а также указание, что лекарственный препарат по истечении срока годности не должно применяться; · условия хранения; · указание, что лекарственный препарат следует хранить в местах, не доступных для детей;
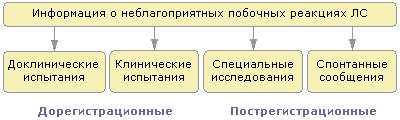
Основные пути решения безопасности лечения. Система фармакологического надзора в мире и Украине. Формулярная система. Медико-юридические и организационные аспекты. роль врача в выявлении побочных реакций лс Ø внешние относительно больного факторы (врач, который проводит фармакотерапию, экология, условия работы и др.). Ø Факторы, связанные с побочным действием препаратов: особенности клинико-фармакологической характеристики ЛС; адекватность выбора препарата; метод применения препарата; взаимодействие ЛС при полипрагмазии. Ø факторы связанные с качеством лс Контролируемые клинические исследования (при изучении новых ЛС).
Все вышеуказанные направления сбора информации имеют свою специфику при непосредственном получении сообщений, исходя из целей исследования, рекомендованных ВОЗ Сейчас в мире, благодаря усилиям ВОЗ, сформировалась и продолжает развиваться система обмена информацией о нежелательных ПД ЛС
Тщательный анализ причин, которые привели к прекращению испытаний, и причин летальности, ставших причиной ограничения или запрещения медицинского применения ЛС. В Украине после провозглашения независимости впервые в истории системы здравоохранения официальным изучением ПД ЛС начало заниматься с 1995 г. подразделение Фармакологического комитета МЗ Украины — Центр побочного действия лекарственных препаратов. Деятельность его была направлена на сбор и анализ, в первую очередь, собственной информации о ПР, которые регистрировалось при проведении официальных клинических испытаний ЛС. Одновременно Центр начал создавать базу спонтанных сообщений о ПД. В 1999 г. Центр был переименован в Отдел фармакологического надзора в составе Государственного фармакологического центра МЗ Украины (правопреемник Фармакологического комитета). В части контроля за ПР/ПД ЛС отдел руководствуется Законом Украины о лекарственных средствах, порядке государственной регистрации/перерегистрации ЛС с учетом норм, которые применяются в международной практике (GCP, ІСН, директив Совета ЕЭС), и т. п. До 2000 г. в ГФЦ МЗ Украины накоплено около 700 спонтанных сообщений о ПР/ПД ЛС, которые поступили от врачей из лечебно-профилактических учреждений и учреждений разных регионов Украины, а также данные, полученные во время клинических испытаний, проводимых под контролем ГФЦ МЗ Украины. Информация о ПР/ПД ЛС поступает в ГФЦ МЗ Украины от: Ø врачей, фармацевтов и медицинских работников независимо от ведомственного подчинения и форм собственности (форма 1,2); Ø производителей/собственников регистрационного удостоверения или их уполномоченных представителей (форма 3); Ø уполномоченных международных организаций (Всемирная организация здравоохранения, Европейское сообщество и т. п.), Ø медицинских источников информации и научных изданий; Ø общественных организаций, которые представляют интересы потребителей лекарственных средств, а также граждан; Ø комиссии по вопросам этики (во время клинических испытаний лекарственных средств); региональных отделений. Центр получает информацию о ПР/ПД ЛС, систематизирует, анализирует в сроки, и готовит информационные сообщения, аналитические обзоры, экспресс-информацию, методические рекомендации, предложения МЗ Украины по изменению инструкции по медицинскому применению и обращению лекарственных средств и т. п. Отчеты о подозреваемых серьезных побочных реакциях ГФЦ МЗ Украины направляет в ВОЗ через бюро ВОЗ в Украине, а копии — в Агентство ЕС.
|