Лечение
При заболеваниях почек лечение состоит из специфического лечения конкретного заболевания и нефропротективного лечения, универсального для всех патологий почек. Специфическое лечение назначается в зависимости от конкретного заболевания. При гломерулонефритах, поражении почек при системных заболеваниях соединительной ткани применяют стероиды, БИАРЛ (болезнь-изменяющие антиревматические лекарства). При инфекционных поражениях почек и мочевыводящих путей — антибиотики. При диабетической нефропатии — коррекция уровня глюкозы крови. Нефропротективное лечение назначается при всех хронических заболеваниях почек и преследует цель замедления прогрессирования почечной недостаточности. Основным в нефропротективном лечении является блокада ренин-ангиотензин-альдостероновой системы за счёт нескольких групп лекарственных препаратов: блокаторов ангиотензин-превращающего фермента, блокаторов рецепторов ангиотензина, антагонистов альдостерона, прямых ингибиторов ренина и др. Важнейшим является лечение, снижающее уровень протеинурии, посредством нормализации внутриклубочковой гипертензии (блокада РААС) и защиты проксимального эпителия от токсического эндоцитоза протеинов (гидрофильные статины и антиоксиданты). Неспецифическим, но важным лечением является антигипертензивная терапия при сопутствующей артериальной гипертензии).
7 Синдрома Альпорта.Определение, Патогенез, Клинические проявления, диагностика, лечение.
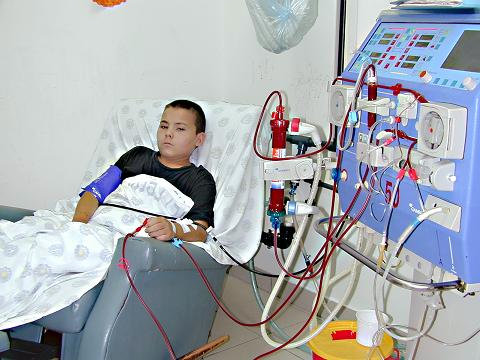
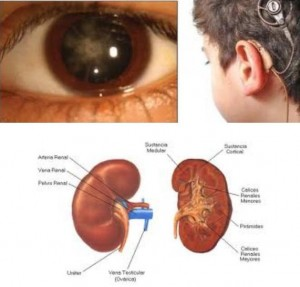
Наследственный нефрит (синдром Альпорта) - генетически детерминированная наследственная неиммунная гломерулопатия, проявляющая гематурией (иногда с протеинурией), прогрессирующим снижением почечных функций с развитием хронической почечной недостаточности, часто сочетающаяся с нейросенсорной глухотой и расстройствами зрения.
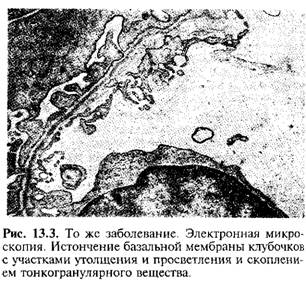
Генетической основой развития наследственного нефрита являются мутации в генах альфа-цепей коллагена IV типа. Известно шесть а-цепей IV коллагена Г типа: гены а5- и а6-цепей (Соl4A5 и Соl4A5) находятся на длинном плече Х-хромосомы в зоне 21-22q; гены а3- и а4-цепей (Соl4A3 и Соl4A4) - на 2-й хромосоме; гены a1- и a2-цепей (Соl4A1 и Соl4A2) - на 13-й хромосоме. В большинстве случаев (80-85%) выявляется Х-сцепленный тип наследования заболевания, связанный с повреждениями гена Соl4A5 в результате делеции, точечных мутаций или нарушений сплайсинга. Результатом мутаций является нарушение процессов сборки коллагена IV типа, приводящее к нарушению его структуры. Коллаген IV типа является одним из основных компонентов гломерулярной базальной мембраны, кохлеарного аппарата и хрусталика глаза, патология которых будет выявляться в клинике наследственного нефрита. Коллаген IV типа, входящий в состав гломерулярной базальной мембраны, состоит в основном из двух а1-цепей (IV) и одной а2-цепи (IV), а также содержит а3, а4, а5-цепи. Наиболее часто при Х-сцепленном наследовании мутация гена Соl4A5 сопровождается отсутствием а3-, а4-, а5- и а6 цепей в структуре коллагена IV типа, а количество о1- и а2-цепей в гломерулярной базальной мембране возрастает. Механизм этого феномена неясен, предполагается, что причиной являются посттранскрипционные изменения мРНК. Отсутствие а3-, а4- и а5-цепей в структуре IV типа коллагена базальных мембран клубочков приводит к их истончению и ломкости на ранних стадиях синдрома Альпорта, что клинически проявляется чаще гематурией (реже гематурией с протеинурией или только протеинурией), снижением слуха и лентиконусом. Дальнейшее прогрессирование заболевания приводит к утолщению и нарушению проницаемости базальных мембран на поздних стадиях заболевания, с разрастанием в них коллагена V и VI типов, проявляющихся в нарастании протеинурии и снижении почечных функций. Характер мутации, лежащей в основе наследственного нефрита, во многом определяет его фенотипическое проявление. При делеции Х-хромосомы с одновременной мутацией генов Соl4A5 и Соl4A6, ответственных за синтез а5- и а6-цепей коллагена IV типа, синдром Альпорта сочетается с лейомиоматозом пищевода и половых органов. По данным исследований при мутации гена Соl4A5, связанной с делецией, отмечаются большая тяжесть патологического процесса, сочетание почечного поражения с экстраренальными проявлениями и ранним развитием хронической почечной недостаточности, по сравнению сточечной мутацией этого гена. Морфологически при электронной микроскопии выявляется истончение и расслоение гломерулярных базальных мембран (особенно lamina densa) и наличие электронно-плотных гранул. Поражение гломерул может быть неоднородным у одного и того же больного, от минимального фокального поражения мезангия до гломерулосклероза. Гломерулит при синдроме Альпорта всегда носит иммунонегативный характер, что отличает его от гломерулонефрита. Характерны развитие атрофии канальцев, лимфогистиоцитарная инфильтрация, наличие " пенистых клеток" с включениями липидов - липофагами. При прогрессировании заболевания выявляется утолщение и выраженная деструкция базальных мембран клубочков.
Выявляются определенные сдвиги в состоянии имунной системы. У больных наследственным нефритом отмечено снижение уровня Ig A и склонность к повышению концентрации IgМ в крови, уровень IgG может быть повышен на ранних стадиях развития заболевания и снижаться на поздних сроках. Возможно, повышение концентрации IgM и G является своеобразной компенсаторной реакцией в ответ на дефицит IgA. Функциональная активность системы Т-лимфоцитов снижена; отмечается избирательное снижение В-лимфоцитов, ответственных за синтез Ig A, нарушается фагоцитарное звено иммунитета, в основном за счет нарушения процессов хемотаксиса и внутриклеточного переваривания в нейтрофилах При исследовании биоптата почек у больных с синдромом Альпорта по данным электронной микроскопии наблюдаются ультраструктурные изменения базальной мембраны клубочка: истончение, нарушение структуры и расщепление гломерулярных базальных мембран с изменением ее толщины и неравномерностью контуров. На ранних стадиях наследственного нефрита дефект определяет истончение и ломкость гломерулярных базальных мембран. Истончение гломерулярных мембран является более благоприятным признаком и чаще встречается у девочек. Более постоянный электронно-микроскопический признак при наследственном нефрите - расщепление базальной мембраны, причем выраженность деструкции ее коррелирует с тяжестью процесса.
|