ЭФФЕКТЫ СОЛЬВАТАЦИИ В ЗНАЧЕНИЯХ ХИМИЧЕСКИХ СДВИГОВ ЯМР 15N АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛОВ
Семенов В.А.,1 Чернышев К.А.2
1 Иркутский институт химии им. А.Е. Фаворского СО РАН, Иркутск, Россия. Аспирант 1г. semenovval@inbox.ru 2 Иркутский институт химии им. А.Е. Фаворского СО РАН, Иркутск, Россия. Молодой учёный. Научный руководитель: Кривдин Л.Б.
Известно, что значения химических сдвигов ЯМР 15N азотсодержащих соединений сильно зависят от природы растворителя. Это особенно актуально при теоретическом расчете химических сдвигов ЯМР 15N, так как ошибки, связанные с пренебрежением влияния среды могут привести к совершенно некорректным выводам о строении изучаемых соединений. Целью настоящего исследования являлось изучение влияния природы растворителя на точность расчетов химических сдвигов ЯМР 15N на примере пиррола и пиридина. Данная работа продолжает наши исследования, посвященные теоретическому расчету химических сдвигов [1] и изучению эффектов координации [2,3]. В настоящей работе влияние неспецифической сольватации было учтено в рамках модели поляризованного континуума (модель 1). Для моделирования специфического взаимодействия, растворитель учитывался в явном виде, то есть путем добавления молекулы растворителя в расчетное пространство (модель 2). Рассчитанные значения химических сдвигов ЯМР 15N и их отклонения от эксперимента (Δ) для пиррола и пиридина в пяти растворителях представлены в таблице 1. Результаты расчета приведены для метода GIAO-B1PW91/TZP, показавшего наилучшее согласие с экспериментом.
Таблица 1. Теоретические а и экспериментальные значения химического сдвига ЯМР 15N пиррола и пиридина в различных растворителях.
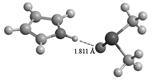
Примечание: а) Теоретические значения химических сдвигов ЯМР 15N в газовой фазе составляют -243.5 м.д. в пирроле и -50.9 м.д. в пиридине. Анализ теоретических значений химических сдвигов ЯМР 15N показывает, что в случае малополярных апротонных растворителей, таких как CCl4 и CHCl3, наблюдается хорошее согласие с экспериментом, причем для учета сольватации достаточно континуальной модели. Отклонения рассчитанных значений от эксперимента не превышает 3 м.д., а введение молекулы растворителя даже несколько ухудшает качество расчета. С другой стороны, в случае метанола, ДМСО и воды приближения PCM уже недостаточно, различия между экспериментом и расчетом превышают 10 м.д. Низкая точность, по-видимому, объясняется образованием слабых межмолекулярных водородных связей между N–H протоном пиррола и неподеленной электронной парой азота пиридина с молекулами растворителей. И действительно, оптимизация геометрии межмолекулярных комплексов исследуемых соединений с одной молекулой растворителя позволила обнаружить такие взаимодействия (рис. 1). Дальнейшие расчеты химических сдвигов ЯМР в указанных комплексах позволили получить теоретические значения химических сдвигов ЯМР 15N лучше согласующиеся с экспериментом (таблица 1).
Рисунок 1. Пространственное строение межмолекулярных комплексов пиррола и пиридина с одной молекулой ДМСО и воды, оптимизированные методом B3LYP/6-311G**.
Таким образом, из результатов данной работы следует, что при расчете химических сдвигов ЯМР 15N азотсодержащих гетероциклов учет эффектов среды необходим, так как пренебрежение может привести к некорректным результатам. При этом, в случае малополярных растворителей достаточным является учет в рамках континуальной модели, а в случаях с полярными протонными растворителями необходим их учет в явном виде. Работа выполнена при финансовой поддержке гранта РФФИ № 11-03-00022
Литература: [1]. Чернышев К. А., Кривдин Л.Б. ЖОрХ, 46, 795-800 (2010) [2]. Чернышев К. А., Ларина Л. И., Чиркина Е. А., Розинов В. Г., Кривдин Л. Б. ЖОрХ, 47, 1823-1828 (2011) [3]. Чернышев К. А., Ларина Л. И., Чиркина Е. А., Розинов В. Г., Кривдин Л. Б. ЖОрХ, 47, 1833-1837 (2011)
|