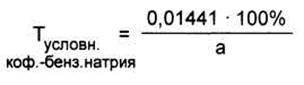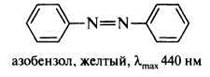ИНФОРМАЦИЯ
1. ОПРЕДЕЛЕНИЕ КОНЦЕНТРАЦИИ СПИРТА В СПИРТО-ВОДНЫХ РАСТВОРАХ В водных растворах этилового спирта линейная зависимость показателя преломления и концентрации наблюдается в пределах до 50-60%. Если необходимо проводить определение крепости спирта в более концентрированных растворах методом рефрактометрии, то следует их предварительно разбавить и при расчетах концентрации учитывать разведение. При определении показателя преломления спирто-водных растворов необходимо на призму рефрактометра наносить не менее 5-7 капель исследуемого раствора и измерять величину немедленно во избежание ошибки, связанной с улетучиванием спирта. Исследование необходимо проводить при температуре 20° С. Если оно осуществляется не при 20° С, следует вносить поправки на температуру. Величины поправок показателя преломления на 1° С представлены в таблице. Если определение проводится при температуре выше 20° С, то поправку прибавляют к найденной величине показателя преломления; если анализ проводится при температуре ниже 20° С, поправку вычитают.
ПРИМЕР 1
Анализу подвергался 40% раствор спирта. Определение показателя преломления проводили при 22° С. Показание рефрактометра – 1,3544. Согласно таблицы, поправка на 1° С для показателя преломления, близкого по величине к полученному (1,35500), равна 2,4 х 10-4 (т. е. 0,00024). Поскольку исследование проводилось при 22° С, то поправка будет составлять 0,00024 х 2 = 0,00048. Для приведения показателя преломления к стандартным 20° С, необходимо определённый показатель (1,3544) сложить с поправкой, умноженной на разность температур (0,00048):
1,3544 + 0,00048 = 1,35488
В таблице находим соответствующую данному показателю преломления концентрацию спирта. Найденной величины показателя преломления (1,35488) в таблице нет; ищем близкое по величине показателю преломления, оно равно 1,35500, что соответствует 40% спирта. Следующее действие: необходимо определить, какая концентрация спирта соответствует разности показателей преломления между взятым нами в таблице (1,35500) и вычисленным значением (1,35488):
1,35500 – 1,35488 = 0,00012.
Продолжаем работать с таблицей: поправка на 1% спирта при 1,35500 равна 4,0 х 10-4. Переводим эту величину в развёрнутый вид 0,0004 и проводим действие:
0,00012 ----------- = 0,3 0,0004
Реальное содержание спирта в исследуемом растворе равно: 40 - 0,3 = 39,7%. Вычитание используется в том случае, если сравниваемое табличное значение больше исчисленного.
Таблица 1. Показатели преломления спирто-водных растворов, концентрация которых выражена в объёмных %.
2. ОПРЕДЕЛЕНИЕ КОНЦЕНТРАЦИИ СПИРТА В СПИРТОВЫХ РАСТВОРАХ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
ИНФОРМАЦИЯ
Для определения концентрации этилового спирта в спиртовых растворах лекарственных препаратов, приготовленных на 70% спирте, по указанной выше причине, проводят разведение обычно 1:2, а приготовленных на 95 % спирте - 1:3. Исключение составляют растворы салициловой кислоты, приготовленные на 70% спирте, которые разводят 2:1 вследствие ограниченной растворимости салициловой кислоты в воде. При этом необходимо учитывать, что при смешивании спирта с водой объем раствора несколько уменьшается, в связи с чем следует вносить поправку к фактору разведения: при смешивании 2 мл спирта с 1 мл воды для получения общего объёма 2 умножают не на 1,5, а на коэффициент 1,47; при смешивании 1 мл спирта с 2 мл воды - на 2,98 (вместо 3); при смешивании 1 мл спирта с 3 мл воды – на 3,93 вместо 4. После соответствующего разведения определяют показатель преломления полученного раствора, вычитают величину показателя преломления, приходящуюся на содержание растворенного препарата (или препаратов) в разбавленном растворе. Если необходимо, вносят поправку на температуру и находят концентрацию спирта в приготовленном растворе. Для установления крепости спирта в лекарственной форме найденное значение концентрации умножают на коэффициент разведения.
Таблица 2. Поправки показателей преломления на содержание салициловой кислоты в разбавленном (2:1) спирто-водном растворе.
ПРИМЕР 2
1. Определение концентрации спирта в 2 % растворе кислоты салициловой, приготовленного на 70% спирте. Раствор анализируется при температуре 20° С. В сухую склянку вносят пипеткой 2 мл раствора кислоты салициловой спиртовой и 1 мл воды, перемешивают и устанавливают показатель преломления полученного раствора (n = 1,3599). Затем из его значения (табл.2) вычитают поправку показателя преломления на содержание салициловой кислоты в разбавленном растворе (0,00188) для 2% раствора и находят показатель преломления спирта в разбавленном растворе:
1,3599 - 0,00188 = 1,35802
Далее вычисляют содержание спирта. По таблице 1 находим, что близкому по значению к найденному экспериментально показателю преломления 1,35700 соответствует 45% спирта. Поправка на 1% спирта равна 4,0 х 10-4 (0,0004), этот показатель также берётся из таблицы 1. Поправка на разность (1,35802 – 1,35700 = 0,00102) равна 0,00102/0,0004 = 2,55 % спирта. Следовательно, содержание спирта в разбавленном 2:1 растворе равно:
45% + 2,55% = 47,55%, а в исходном растворе: 47,55 x 1,47 = 69,90%
Возможна также ситуация, когда в спирто-водном растворе титриметрическими методами будет найдено несколько иное содержание растворённого компонента (в данном случае салициловой кислоты), отличное от исходной прописи. В этом случае поправку, приведённую для 1% раствора вещества, умножают на фактически найденное процентное содержание и затем вычитают её из найденной величины показателя преломления ПРИМЕР 3 В 2% растворе салициловой кислоты найдено фактическое её содержание 1,85%. В этом случае поправка показателя преломления по содержанию салициловой кислоты будет подсчитана так:
0,00094 х 1,85 = 0,001739
Этот показатель надо будет отнять из найденного показателя преломления 2% раствора салициловой кислоты вместо 0,00188.
ПРИМЕР 4 2. Определение концентрации спирта в 1 - 4 % растворах борной кислоты, приготовленных на 70% спирте. Для определения концентрации спирта к 1 мл лекарственной формы добавляют 2 мл воды очищенной, перемешивают и измеряют показатель преломления приготовленного разведения. Далее вычитают поправку на содержание борной кислоты (табл. 3). Если необходимо, вносят поправку на температуру и устанавливают содержание спирта, учитывая коэффициент разведения 2,98.
Количественное определение лекарственных препаратов в спиртовых растворах рекомендуется проводить объемно-аналитическим методом, так как рефрактометрические методы требуют приготовления в качестве контроля (n0) раствора спирта точно такой же концентрации, как и в исследуемом растворе, что усложняет анализ.
ИНФОРМАЦИЯ
Содержание в лекарственных смесях веществ, близких по химическому строению и свойствам (сульфаниламиды, соли галогеноводородных кислот и др.) затрудняет их раздельное определение общепринятыми титриметрическими методами. В этом случае как исключение допускается применять средний ориентировочный титр (СОТ) для определения суммы веществ. Иногда этот титр называют суммарным. СОТ – это масса смеси определяемых веществ в граммах, соответствующая 1 мл титранта. Кулешова М. И. с соавторами предлагает его определять как результат отношения суммарного количества определяемых веществ в массе навески (а), взятой для анализа, в граммах к суммарному теоретическому объёму (А) титрованного раствора, необходимому для их титрования, в мл, то есть: а Тср = -------; А СОТ может быть рассчитан различными способами.
ПРИМЕР 1 При суммарном титровании хлоридов натрия, калия и кальция 0,1 моль/л раствором серебра нитрата в растворе Рингера средний титр (Тср) вычисляется следующим образом: в 1 мл раствора, взятом для анализа, содержится: натрия хлорида 0,009 г, калия и кальция хлоридов по 0,0002 г. Титр натрия хлорида - 0,005844 г, калия хлорида — 0,007456 г, кальция хлорида — 0,01095 г.
0,009 1) на натрия хлорид - -------------=1,54 мл; 0,005844
0,0002 2) на калия хлорид - ------------ = 0,027 мл; 0,007456
0,0002 0,01095
С использованием этих данных получим:
0,009 + 0,0002 + 0,0002 0,0094 Tcp = -------------------------------- = -----------= 0,00593 1,54 + 0,027 + 0,018 1,585
В случае, если титруемые совместно вещества находятся в ЛФ в одинаковом количестве, применяется упрощённая формула, позволяющая рассчитывать СОТ: ПРИМЕР 2
1) Rp: Natrii bromidi Kalii bromidi ana 4,0 Aq. purific. 200 ml # Сумму калия и натрия бромида определяют меркуриметрически, рассчитывая по СОТ. Титр для натрия бромида при использовании 0,1 N раствора Hg(NO3)2 = 0,01029; титр для калия бромида в тех же условиях = 0,0119. СОТ равен:
0,01029 + 0,0119 Тср =-----------------------= 0,011095
2) Упрощённый вариант для одновременного определения двух компонентов, которые содержатся в ЛФ в неодинаковом количестве:
Б + В Тср =---------------, где Б/Т1 + В/Т2
Б - прописанная масса первого компонента в прописи; В - прописанная масса второго компонента в прописи; Т1 – титр первого компонента; Т2 – титр второго компонента.
ПРИМЕР 3
Rp: Natrii bromidi 4,0 Kalii bromidi 5,0 Aq. purific. 200 ml #
4 + 5 9 Тср =-------------------------- = -------------------------= 0,01112 4/0,01029 + 5/0,0119 388,7269 + 420,1681
3) Ещё один вариант для расчёта СОТ:
Т1 х Б + Т2 х В Тср =--------------------, где Б + В
Б - прописанная масса первого компонента в прописи; В - прописанная масса второго компонента в прописи; Т1 – титр первого компонента; Т2 – титр второго компонента.
УСЛОВНЫЙ ТИТР ИНФОРМАЦИЯ
Некоторые лекарственные вещества представляют собой комплексные соединения, состоящие из двух веществ (кофеин-бензоат натрия, эуфиллин, темисал, протаргол и др.). Такие соединения в лекарственных смесях можно определять по входящим в них компонентам, содержание которых согласно требованиям ГФ и НД должно быть в строго определенных пределах. Например, кофеин-бензоат натрия в экспресс-анализе чаще анализируют по бензоату натрия, которого в препарате должно быть от 58 до 62%. Если пользоваться титром 0,01441 г/мл, исходя из М. м. натрия бензоата (144,1 г/моль) и титрантом - 0,1 моль/л раствором кислоты хлороводородной, то в результате получим содержание натрия бензоата в лекарственной форме. Для пересчета на кофеин-бензоат натрия полученный результат нужно дополнительно поделить на фактическое содержание (массовую долю) натрия бензоата в кофеин-бензоате натрия. Для упрощения расчётов, можно использовать условный титр,пересчитанный на препарат. Для кофеина-бензоата натрия его определяют по формуле:
где а – содержание натрия бензоата в данном образце кофеина-бензоата натрия (в %); 0,01441 - масса натрия бензоата (в г), соответствующая 1 мл 0,1 моль/л раствора кислоты хлороводородной. Величина Тусловн. может значительно колебаться. При содержании в кофеине-бензоате натрия бензоата 58% Тусловн = 0,02484,а при 62% Тусловн = 0,02324.Поэтому для определения Тусловн. необходимо знать реальное содержание натрия бензоата в препарате. Если таких данных нет, расчеты ведут по среднему пределy содержания данного компонента в препарате. Его рассчитывают так: складывают минимальное значение содержания компонента в препарате с максимальным и делят на 2. В нашем случае: (58 + 62)/2 =60; при этом Тусловн = 0,02402.
ПРИМЕР 4
1. Определение кофеина-бензоата натрия ацидиметрически (по натрия бензоату). fэкв натрия бензоата равен 1. М. м. = 144,11 г/моль
0,1 х 144,11 Т(натрия бензоата по 0,1 моль/л хлороводородной кислоте) = ------------------= 0,01441 г/мл.
Для расчёта условного титра необходимо знать процентное содержание натрия бензоата в ЛП. По ФС, он должен находиться в препарате в пределах от 58 до 62%. Так как у нас нет предварительной информации о точном содержании натрия бензоата в конкретной партии, расчёт ведём по среднему пределу содержания данного компонента (см. выше):
0,01441 х 100% Туслов =--------------------= 0,02402 г/мл. 60%
2. Определение препарата кофеин-бензоат натрия по кофеину методом йодометрии. fэкв кофеина ¼; М. м. = 194,19 г/моль.
194,19/4 х 0,1 Т(кофеина по 0,1 моль/л йоду)= -------------------- = 0,004855 г/моль.
По ФС содержание кофеина в ЛП кофеин-натрия бензоат должно находиться в пределах от 38 до 40%. Вычисляем средний предел содержания компонента (см. выше) – (38 + 40)/2=39, подставляем в формулу для расчёта условного титра: 0,004855 х 100% Туслов =--------------------= 0,01245 г/мл. 39% ЗАДАЧА
По полученным экспериментальным данным рассчитать количественное содержание кофеина-бензоата натрия по кофеину методом обратной йодометрии, если на анализ взяли 0,3102 г препарата, растворили в мерной колбе на 200 мл и для анализа взяли 100 мл; при определении на титрование пошло 11,2 мл 0,1 моль/л раствора натрия тиосульфата, в контрольном опыте – 23,8 мл, К = 1,01.
РЕШЕНИЕ
Используем формулу для расчёта с учётом разведения:
0,01245 х (23,8-11,2) х 1,01 х 200 х 100% С%=------------------------------------------------------ =102,15% 0,3102 х 100
ПРИМЕР 5
Эуфиллин (аминофиллин). Определение препарата по компоненту этилендиамину методом ацидиметрии. fэкв этилендиамина ½; М. м. = 60,1 г/моль.
60,1/2 х 0,05 Т(этилендиамина по 0,05 моль/л HCl) =------------------= 0,0015025 г/моль.
По ФС содержание этилендиамина в препарате должно быть в пределах от 14 до 18%. В данной серии этилендиамина 15%.
0,0015025 х 100% Туслов =-----------------------= 0,010017 г/мл 15%
Рассчитать содержание эуфиллина, если на титрование 2 мл 0,5% раствора препарата израсходовано 1,04 мл 0,05 моль/л раствора хлороводородной кислоты, К =0,98. Содержание этилендиамина в данной партии эуфиллина 15%. С использованием условного титра формула для расчёта выглядит так:
1,04 х 0,010017 х 0,98 х 100% С% =---------------------------------------= 0,51%
Без использования условного титра формула для расчёта выглядит так:
1,04 х 0,0015025 х 0,98 х 100% х 100% С% =--------------------------------------------------= 0,51% 2 х 15%
ИНФОРМАЦИЯ Одним из общих свойств молекул является способность к избирательному поглощению электромагнитного излучения, что положено в основу определения строения и идентификации веществ. Человеческий глаз может воспринимать только малую часть волнового диапазона. Механизмы взаимодействия электромагнитного излучения и вещества значительно отличаются в разных частях спектра, но в любом случае происходит поглощение определённого количества энергии. Поглощение в ультрафиолетовой и видимой области связано с поглощением электромагнитного излучения электронами.
УЛЬТРАФИОЛЕТОВАЯ СПЕКТРОСКОПИЯ Электронные спектры молекул проявляются в ультрафиолетовой и видимой областях спектра, т. е. в области электромагнитных излучений с длиной волны от 100 до 800 нм. Спектральные свойства молекул зависят от типа содержащихся в них валентных электронов. Электроны, образующие ординарную связь, называются σ-электронами (в соответствии с названиями молекулярных орбиталей). Характеристические функции и плотности заряда этих электронов враща-тельно-симметричны по отношению к валентной связи. Электроны, образующие двойную связь, называются π-электронами. Характеристические функции и плотности их заряда имеют узловую плотность колебания, проходящую через валентную связь. В молекулах, содержащих атомы азота, серы, кислорода и других, имеются неспаренные или несвязанные электроны, которые называются n-электронами. Поглощение световой энергии органическими соединениями в видимой и ультрафиолетовой областях спектра связано с переходом σ-, π- и n-электронов из основного состояния в состояние с более высокой энергией. Эти возбужденные состояния соответствуют молекулярным орбиталям, которые называют обычно разрыхляющими или анти-связывающими орбиталями. Разрыхляющие орбитали, соответствующие σ-связям, называются σ*-орбиталями, а π-связям — π*-орбиталями. Так как n-электроны не образуют связей, то соответствующих им разрыхляющих орбиталей не существует. Имеются следующие типы электронных переходов (→), происходящих в УФ- и видимой частях спектра: σ→σ*, п →σ*; п →π* и π→π *. Энергия, необходимая для перехода σ→σ*, очень велика, поэтому соединения, у которых все валентные электроны участвуют в образовании ординарных связей, (насыщенные углеводороды), не поглощают света в ближней ультрафиолетовой области. Для σ→σ* перехода необходима энергия, которой обладают очень короткие волны дальней (вакуумной) ультрафиолетовой области, т. е. волны <200 нм. Циклические парафины поглощают при более длинных волнах, чем соответствующие им соединения с открытой цепью, так как из-за напряженности циклических молекул возникает некоторая ненасыщенность, п →σ* переходы требуют меньше энергии, чем переходы σ→σ*, поэтому молекулы с неспаренными электронами, как правило, имеют максимум поглощения в ближней УФ-области. Переходы на разрыхляющие π*-орбитали обусловлены ненасыщенными участками молекулы; для них требуется еще меньшая энергия и наблюдаются они при больших длинах волн, вполне доступных для определения на обычных спектрофотометрах. Следовательно, существенными элементами, обусловливающими наличие электронных спектров органических молекул, являются кратная связь и неподеленная пара электронов. Их наличием в молекуле и их многочисленными сочетаниями объясняется вся совокупность электронных спектров органических соединений в ближней ультрафиолетовой области.
ИСПОЛЬЗУЕМЫЕ МЕТОДЫ И АППАРАТУРА Из физико-химических методов для количественного определения многих лекарственных препаратов в ГФХ и последующих ГФ наиболее широко представлены фотометрические методы: спектрофотометрия и фотоколориметрия. Эти методы отличаются простотой выполнения анализа, достаточной точностью, высокой чувствительностью и небольшой затратой исследуемого вещества. Фотоколориметрический метод основан на измерении поглощения света не строго монохроматического излучения окрашенными соединениями в видимой области спектра. Если исследуемые соединения бесцветны, их переводят в окрашенные соединения путем взаимодействия с различными реактивами. В этом случае окрашенные соединения в большинстве своем являются комплексными или внутрикомплексными соединениями. Последние должны быть прочными, иметь постоянный состав, высокую интенсивность окраски и т. д. Затем определяют оптическую плотность окрашенного раствора исследуемого вещества. Это делается или с помощью спектрофотометров в видимой области спектра, либо чаще всего с помощью фотоэлектроколориметров. У последних имеется набор светофильтров, с помощью которых можно выделить более узкий интервал длин волн не монохроматического излучения. Основной принцип работы всех систем электрофотоколориметров заключается в том, что световой поток определенного интервала длин волн, прошедший через кювету с окрашенным раствором или растворителем, попадает на фотоэлемент, который превращает световую энергию в электрическую, измеряемую гальванометром. Спектрофотометрический метод основан на измерении поглощения света определенной длины волны (монохроматического излучения) точнее, очень узкого интервала длин волн (1-2 нм). Эти измерения поглощения света осуществляются при помощи спектрофотометров, в которых используется всегда монохроматический поток световой энергии, получаемый посредством оптической системы — монохроматора. Спектрофотометрический метод анализа имеет некоторые преимущества перед фотоколориметрическим. Основные из них следующие: 1. Значительное увеличение чувствительности и точностиколичественного определения вследствие возможности работы в узкой области оптимального светопоглощения. 2. Метод спектрофотометрии может использоваться не только для анализа индивидуального вещества, но и для анализа смесей, содержащих несколько поглощающих и не взаимодействующих химически друг с другом компонентов. 3. Спектрофотометры позволяют работать не только с окрашенными растворами, которые поглощают свет в видимой части спектра (400-760 нм), но и с «бесцветными» для глаза растворами, которые поглощают излучение в ультрафиолетовой (200-400 нм) или ближней инфракрасной (760-1100 нм; 760-2500 нм) областях спектра. 4. Спектрофотометрические методы дают возможность определять константы диссоциации различных веществ, состав комплексных соединений и т. п. Таким образом, возможности спектрофотометрического метода анализа значительно шире, чем фотоколориметрического.
ХРОМОФОРЫ И АУКСОХРОМЫ Еще более ста лет назад окраску веществ связывали с наличием в их структуре так называемых хромофорных групп, к которым относятся некоторые ненасыщенные группировки, например, двойные связи С=С, С=О, C=N, N=N, N=O, ароматические фрагменты. Изолированные хромофоры имеют полосы поглощения в электронном спектре в дальней ультрафиолетовой области (165-200 нм) и являются прозрачными в видимой области спектра. Сопряжение одного хромофора с другим вызывает сдвиг полос поглощения в сторону больших длин волн с одновременным увеличением их интенсивности. Окрашенные вещества поглощают в видимой области спектра (400-800 нм). Очевидно, что такие соединения должны иметь в своей структуре длинную цепь сопряжения. Типичным примером окрашенных веществ служат азосоединения, характеризующиеся наличием в структуре в качестве главного хромофора фрагмента азобензола. Сопряженная система азобензола включает два бензольных кольца и азогруппу:
Различные азосоединения в зависимости от длины сопряженной системы могут быть окрашены в желтый, оранжевый, красный, синий и зеленый цвета. Изменению и углублению окраски способствует наличие в структуре ауксохромов — атомов или групп атомов, вступающих в р, π - и π, π-сопряжение с π-электронной системой главного хромофора. Наиболее интенсивную окраску имеют те соединения, в которых с главным хромофором сопряжены одновременно электронодонорные и электроноакцепторные группы, находящиеся в пара- или орто-положении по отношению друг к другу.
КОНТРОЛЬНАЯ РАБОТА № 3
Контрольная работа №3 содержит вопросы из раздела: - лекарственные средства органической природы: Гетероциклические соединения природного и синтетического происхождения
|
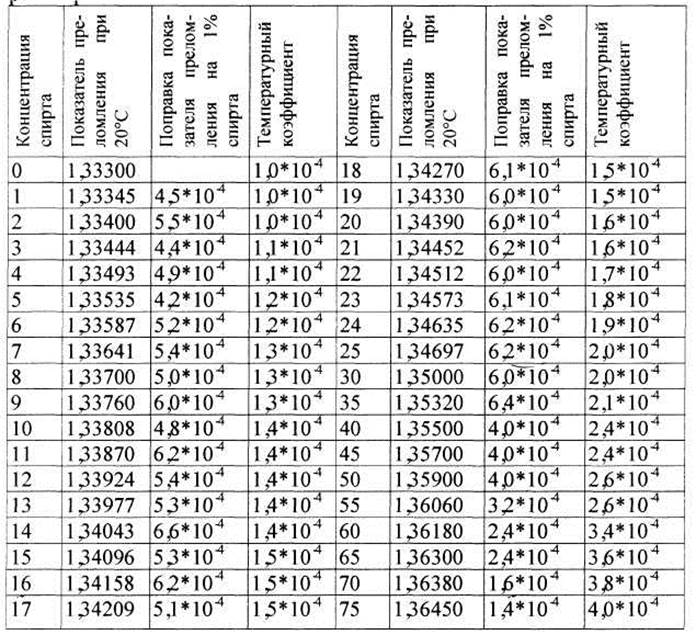
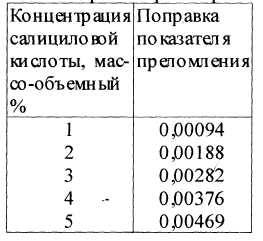
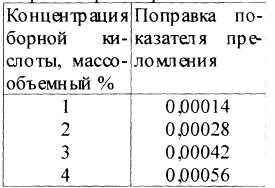 Таблица 3. Поправка показателей преломления на содержание борной кислоты в разбавленном 1:2 спирто-водном растворе.
Таблица 3. Поправка показателей преломления на содержание борной кислоты в разбавленном 1:2 спирто-водном растворе.